
記住我
Single-cell RNA-sequencing (scRNA-seq) technologies allow for the detailed analyses of the transcriptome of every single cell in each tissue or organ. This enhanced and unprecedented resolution can better classify cells and cell states as well as identify cell-specific changes in gene expression. This technology dramatically increases the detail, accuracy, and resolution of our map of the living organism [1, 2]. Indeed, in just a few years after its introduction, scRNA-seq technologies have been widely implemented, led to major breakthroughs, and improved our understanding of the normal and pathologic conditions of several tissues and organs [3]. Technological and computational breakthroughs have facilitated an exponential increase in the number of cells profiled, now having the capability to capture information from over a million cells per study [4, 5]. Thus, scRNA-seq is being used in large-scale efforts to provide a high-resolution map of every cell in the human body [6].
The lung is one of the most complex organs of the human body with over forty cell types specializing in gas exchange, surfactant production, and protection against pathogens and harmful pollution [7,8,9]. In recent years, advanced parenchymal lung disease has emerged as a major cause of mortality and morbidity. Chronic Obstructive Pulmonary Disease (COPD), most commonly caused by exposure to cigarette smoke but also indoor pollution [10] is now considered the 3rd leading cause of death in the world [11]. Fibrotic interstitial lung disease (fILDs), which are a heterogeneous group of lung disorders that cause progressive scarring of the lung parenchyma and are associated with substantial morbidity and mortality [12,13,14]. Of the fILDs, idiopathic pulmonary fibrosis (IPF) is the most lethal, with a median survival of two to five years after diagnosis. Acute respiratory distress syndrome (ARDS) is a highly lethal respiratory failure syndrome and has been under the public radar in the past, but the COVID-19 pandemic has brought it to immediate attention as it is the major cause of death in severe COVID-19. After recovering from the immediate illness, many patients have experienced long-standing symptoms, known as long-COVID [15], and it is estimated that about 15–20% of COVID-19 patients develop a transient pulmonary fibrosis-like disease one year after the infection [16], with the percentage developing long-term sequela still unknown.
In this review, we describe the insights derived by applying single-cell profiling technologies to parenchymal lung disease with a specific focus on fILD and COVID-19, as those have been studied most. So far, scRNA-seq technologies have been applied to samples obtained to close to 200 patients with fILD and COVID-19 as well as healthy controls. Because of the vastness of the data, we chose to organize the discussion based on distinct cell populations, covering the discovery of novel cell populations, the description of known cell populations, and the changes in gene expression within distinct cell populations. All the methodological information of the studies reviewed in this article, including samples, technology, and main findings, is summarized in Table 1. Figure 1 summarizes the perspectives developed in this paper regarding the pathophysiology of progressive lung disease in fILDs and COVID-19.
Table 1 Summary of single-cell RNA-sequencing studies of healthy and fibrotic human lung tissueFig. 1
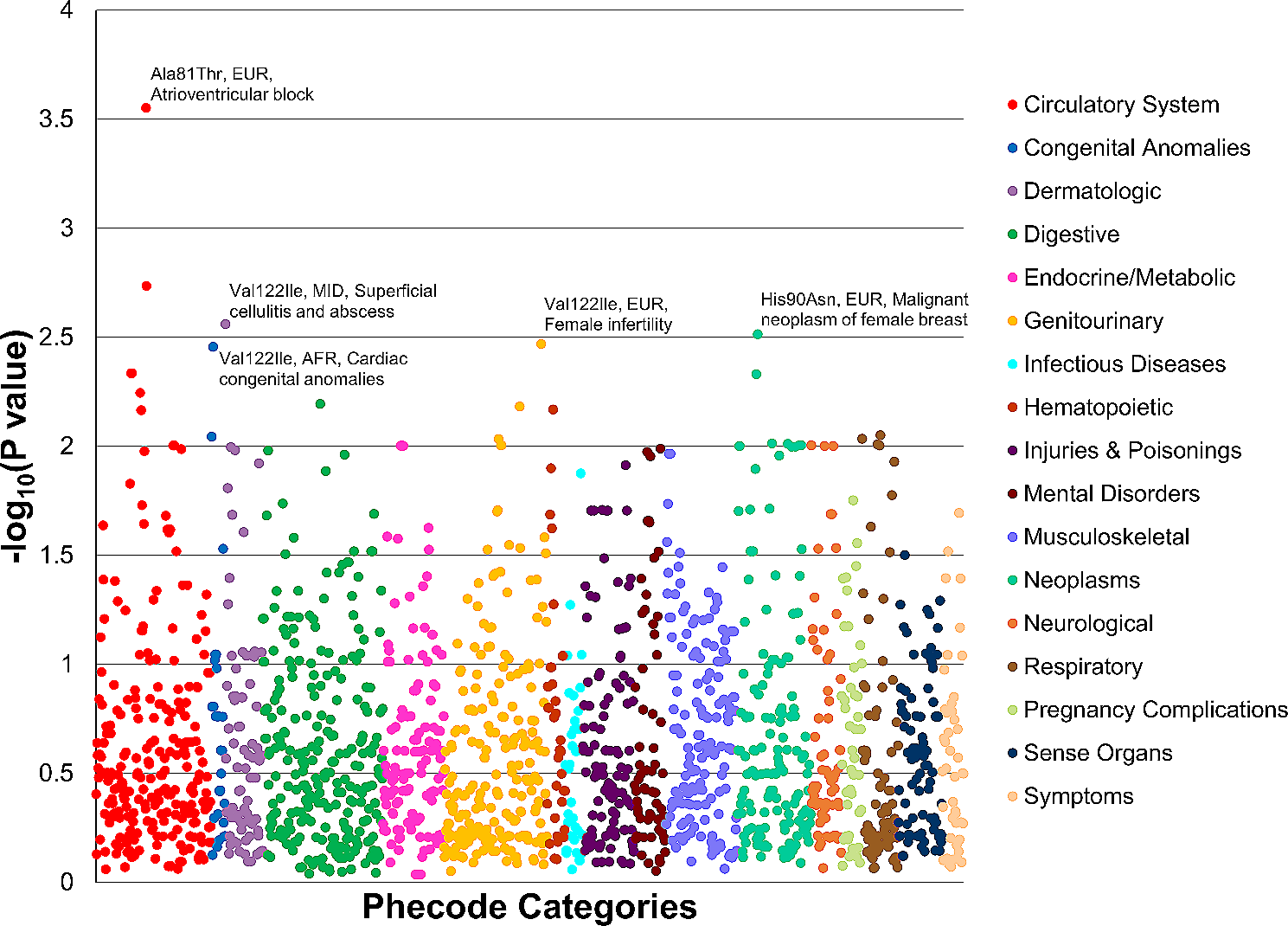
Representative figure of the distal alveolus for healthy control, IPF, and COVID-19 patient samples. Distal aberrant changes in early lungs. A Distal airway and alveolus from a healthy control lung. Several epithelial cell types can be found in the bronchiole epithelium, including club cell, ciliated cell, and basal cell. Basal cells are the airway epithelial progenitor. The alveoli are located in the respiratory bronchioles as scattered outpockets, extending from their lumens. Each lung contains approximately 150 million alveoli providing a surface of gas exchange of forty square meters. Each alveolus is wrapped in a fine mesh of capillaries covering about 70% of its area and composed of endothelial and venule cells. There are three major types of alveolar cells. Two types of pneumocytes or pneumonocytes are known as type 1 (AT1) and type 2 (AT2) cells that are found in the alveolar wall, and a large phagocytic cell known as an alveolar macrophage that moves about in the lumens of the alveoli and the connective tissue. AT1 cells are thin and are involved in the process of gas exchange between the alveoli and blood. AT2 cells are cuboidal and produce surfactant, a lipoprotein that reduced the alveolar tension. AT2 cells also serve as the local alveolar epithelial progenitor. The alveolar fibroblasts are located in the interstitial compartment and are the main source of ECM proteins like collagen and elastin that allow the alveoli to stretch when they fill with air during inhalation. B In IPF, repeated injuries of a senescent alveolar epithelium and bronchiole can lead to the loss of alveolar resident AT1 and AT2 cells, and an increase in airway epithelial cells, such as club cells, basal cells, ciliated cells suggesting “proximalization” of the distal lung. In the vascular compartment, there is a loss in alveolar capillary cells corresponding to gCAP and aerocytes with a concomitant ectopic increase in venous ECs (COL15A1pos) in the lung parenchyma. Immunohistochemistry (IHC) confirmed their presence in fibrotic and bronchiolized areas in IPF lungs paralleling the proximalization of the distal lung observed in the epithelium. In addition to changes in these well-described lung resident cell populations, a new subpopulation of cells distinct was identified and called aberrant basaloid cells. Regarding the immune cells, monocyte-derived macrophages also contribute to fibrosis through increased recruitment and extravasation of these cells, as well as their secreted molecules that also lead to a profibrotic environment. The change of the cellular composition of the epithelial, immune, and endothelial compartments lead to an abnormal secretion of profibrotic mediators such as TGF-β and MMP7 and to the differentiation of alveolar fibroblasts into myofibroblasts. These cells are postulated as the main source of aberrant production of ECM deposition in the interstitial space, ultimately leading to the destruction of the alveolar space and to fibrosis of the lung. C During end-stage COVID-19 lung infection, a similar but different process occurs. SARS-Cov-2 preferentially infects AT2 and AT1 cells, monocyte-derived macrophages, and endothelial cells, causing apoptosis of AT1, AT2, and endothelial cells. To compensate for ATI and ATII cell loss, KRT5 + basal progenitors proliferate and migrate into the alveolus. The injury of the alveolar epithelium also leads to the differentiation of alveolar fibroblasts into myofibroblasts. These cells may contribute to the deposition of the extracellular proteins in the interstitial space. It is unclear what happens to endothelial cell proportions, though it has been postulated that these cells decrease. Viral infection load also correlates directly with gCAPs and aerocytes proportions. There is also an increase in the recruitment and extravasation of macrophages as well as an increase in activated T cells
Epithelial cellsInjury to alveolar epithelial cells is central to all parenchymal lung diseases. In pulmonary fibrosis, loss of the following cells occurs: alveolar type I (AT1) cells, which are flat cells that make up much of the alveolar cells and carry out gas exchange, and alveolar type II cells (AT2), which secrete surfactant, regulate alveolar fluid, and serve as local progenitor cells and has been described long ago [17, 18].
Single-cell transcriptomic analysis confirms this notion. All scRNA-seq studies of fibrotic human lungs revealed loss of alveolar resident AT1 and AT2 cells and an increase in airway epithelial cells, such as club cells, basal cells, ciliated cells [19,20,21,22], suggesting “proximalization” of the distal lung. Airway basal cells, which are epithelial progenitor cells characterized by the expression of P63, KRT17, and KRT5 and are capable of differentiating into any type of airway cells [23] are also increased. Airway basal cells have been previously reported to be increased in the IPF lung [24], and their presence in the lavage fluid of patients with IPF indicates the worst prognosis [25]. In addition to changes in these well-described lung resident cell populations, our group identified a subpopulation of cells distinct from any previously described in the lung. We used the term aberrant basaloid cells to describe them, because they expressed common basal cell markers, such as P63, LAMB3, and KRT17, but did not express KRT5 or KRT15. The aberrant basaloid cells have several intriguing features. They are epithelial in nature but express mesenchymal markers, including COL1A1, TNC, HMGA2, CDH2, which suggests a partial epithelial–mesenchymal transition [20]. Moreover, these cells express senescence-related genes, such as CDKN1A, CDKN2A, CCND1, CCND2, MDM2 as well as GDF15, which has recently been proposed as an epithelial cell senescence marker [26, 27]. Aberrant basaloid cells also express numerous genes previously implicated in IPF, suggesting that they may play a key role in pulmonary fibrosis. MMP7 (matrilysin), a matrix metalloproteinase that cleaves proteoglycans, fibronectin, elastin, and casein, is highly expressed in aberrant basaloid cells. MMP7 is important in pulmonary fibrosis. Mice lacking MMP7 are relatively protected from bleomycin-induced fibrosis [28], and increased blood levels of MMP7 have been shown to distinguish IPF patients from controls and other diseases [29] and are the most validated markers of increased mortality in IPF [30, 31]. Aberrant basaloid cells also express AVB6, an epithelial-restricted integrin that is not expressed in the healthy adult human lung and is known to drive fibrosis by activating TGF-β by binding its latency-associated peptide (LAP) of TGF-β and is an emerging therapeutic target for pulmonary fibrosis [32]. Additional evidence of the potential role of these cells in driving fibrosis is that in IPF lungs, these cells consistently localize to the epithelial layer covering myofibroblast foci [20]. The cells are found in other datasets and simultaneously described by Habermann et al. [21].
Single-cell analyses of COVID-19 lung injury were mostly limited to patients with advanced COVID-19 lung disease with samples obtained at lung transplantation or postmortem [33,34,35,36]. The papers reported similar losses of loss AT2 and AT1 cells with marked transcriptional changes in the remaining cells. AT2 exhibited decreased expression of genes encoding for surfactants, such as SFTPC or SFTPA [33,34,35], similar to what is seen in pulmonary fibrosis. They also exhibited high expression of genes associated with host viral response, including those for programmed cell death such as STAT, TNFSF10, inflammation like IRF3, adaptive and innate immune response such as IFI16 or HLA-A. AT2 cells showed a decrease of ETV5, a transcription factor required to maintain AT2 identity. CAV1, a marker of late AT1 maturation, was also expressed at significantly lower levels in the COVID-19 lung. In addition, AT2 showed enrichment for TNF and HIF1a signaling and had an overrepresentation of 53 pathways that suggested an arrest of the cellular division and a blockade in this transitional state that explains, at least partially, the decrease of AT1 cells. Recent studies have shown that inflammation can induce a cell state that is characterized by failure to fully transition to AT1 cells; this phenomenon has been coined “damage-associated transient progenitors” (DATPs), “alveolar differentiation intermediate” (ADI), or “pre-AT1 transitional cell state” (PATS) [37,38,39]. This program signature (KRT8,CLDN4,CDKN1A) is induced during lung injury [40] and is increased in COVID-19 pneumocytes [35]. These cells are distinct from aberrant basaloid cells, previously described in the IPF lung, but aberrant basaloid cells have been reported in the lungs of two patients with end-stage COVID-19 [36]. The existence of these cells must be confirmed in a larger cohort but could suggest the development of irreversible fILD, for which transplantation is likely the only therapeutic option.
The presence of tuft cells, a rare subset of epithelial cells involved in airway inflammation and intestinal tissue regeneration implicated in lung response to viral pneumonia [41], has also been postulated to be in the lungs of patients with COVID-19 pneumonia [33]. Tuft cells express TAS2 receptors, which have been shown to attenuate allergic airway inflammation [42], DLKC1, and POU2F3 and emerge ectopically in the lung after influenza virus infection [43], where they may contribute to dysplastic remodeling. In COVID-19, Melms et al. identify tuft cell-like subpopulations in the upper airway, which were ectopically present in the parenchyma. Further studies are needed to confirm the existence of these cells in the COVID-19 infected lung and to elucidate their implication in lung remodeling.
Immune cellsSingle-cell studies of advanced parenchymal lung disease are dominated by changes in macrophages populations. In fILDs, Reyfman et al. described a profibrotic alveolar macrophage subtype seen in patients with IPF, systemic sclerosis-associated ILD, myositis-associated ILD, and hypersensitivity pneumonitis [19]. Clustering of lung macrophage single-cell data revealed distinct subclusters that were specific to patients with lung fibrosis. In particular, genes that were differentially expressed in monocyte-derived alveolar macrophages included CHI3L1, MARCKS2, IL1RN, PLA2G7, MMP9, and SPP1. To confirm these findings, the authors performed in situ RNA hybridization to localize these putative profibrotic macrophage subtypes using SPP1 and CHI3L1 and confirmed alveolar macrophage heterogeneity in patients with pulmonary fibrosis [19]. Morse et al. sequencing nine lung samples (three healthy controls, three IPF lower lobes, and three IPF upper lobes), reported three distinct macrophage subtypes in both normal and fibrotic lungs, including (1) monocyte markers-expressing, (2) FABP4hi, and (3) SPP1hi macrophages [44]. SPP1hi macrophages were significantly increased in the lower lobes of patients with IPF, where fibrosis was more pronounced and expressed MERTK, LGMN, and SIGLEC10. We confirmed this finding and further applied archetypal analysis to three different subtypes: classical monocytes, profibrotic IPF macrophages, and control-enriched inflammatory macrophage subtype [20]. Building on these findings of what they term “IPF-expanded macrophages,” Ayaub et al. conducted both CyTOF and flow cytometry analyses in addition to re-analysis of aforementioned single-cell datasets [45]. Using flow cytometry, they discovered that CD84 + CD36 + macrophages were expanded in IPF compared to control and COPD lungs and that this subpopulation aligns with previously described macrophages enriched in IPF, providing a cell surface marker protein-based validation of the scRNA-seq classification.
Within the lungs of COVID-19 patients, Bharat et al. analyzed lung explants from patients with end-stage COVID-19 lung disease and identified a profibrotic macrophage subpopulation expressing similar markers as those found in pulmonary fibrosis patients [36]. The profibrotic monocyte-derived alveolar macrophages expressed SPP1, ILRN, MMP9, CHI3L1, and PLA2G7. Conversely, tissue-resident macrophages were nearly depleted in COVID-19 patients as compared with donor lungs from healthy controls. There were other subpopulations of macrophages that were specifically enriched in COVID-19 patients. A subset of COVID-19 samples had a subtype of macrophages expressing osteoclast-like markers, potentially in calcified and necrotic lung tissue. Furthermore, an inflammatory macrophage subset highly expressing genes involved in lipid and iron metabolism, immune signaling, and cell motility were found in a lung explant but not postmortem biopsies of COVID-19 patients. Overall, the authors found similar profibrotic macrophages in COVID-19 as in pulmonary fibrosis patients suggesting a role for this cell population in sustaining fibrosis.
Melms et al. [36] described aberrant activation of myeloid cells including monocytes, monocyte-derived macrophages, and resident alveolar macrophages which were more frequent in COVID-19 lungs. monocyte-derived macrophages exhibited increased expression of CTSB, CTSD, CTSZ, PSAP) and alveolar macrophages exhibited reduced expression AXL, a regulator of efferocytosis (i.e., clearance of apoptotic cells). Rendeiro et al. [34] found that around 8% of macrophages were positive for S protein. These macrophages exhibited higher expression of apoptosis and inflammatory markers like cleaved CASP3, pSTAT3, KIT, and IL6 but were negative for complement proteins C5b-C9. Moreover, CD14 + CD16 + CD206 + CD163 + CD123 + interstitial macrophages were increased in lungs with late COVID-19 compared to healthy lungs. Monocytes in early COVID-19 displayed the highest expression of IL1B, which may recruit neutrophils. Indeed, in patients with COVID-19, the authors observed increased interactions between macrophages and neutrophils as well as between neutrophils and macrophages. However, inter-macrophage interactions were decreased in COVID-19. Hasan et al. further substantiated this claim in their meta-analysis, finding that inflammatory monocytes and macrophages were significantly increased in severe and deceased COVID-19 patients compared to those who had milder forms of the disease [46]. While the analyses are limited by small numbers of tissues, it suggests that at least in refractory cases, monocyte-derived macrophages with features similar to “profibrotic macrophages” in pulmonary fibrosis accumulate in COVID-19 lungs.
Endothelial cellsDysfunctional endothelial cells have been previously linked to fibrotic lung disease; however, in vitro studies have been limited by difficulty with endothelial cell cultures. Thus, endothelial cells were not previously studied in earnest in the context of fibrotic lung disease until the advent of single-cell RNA-sequencing. Recently, we created a comprehensive atlas of endothelial cells in the lung [47]. We conducted a meta-analysis of lung single-cell RNA-sequencing datasets, selecting for endothelial cells (ECs) using canonical markers, including PECAM1, CDH5, CLDN5, and ERG. Endothelial cells in the lungs were broadly categorized into two subtypes: vascular (example markers include ENG, PCDH17, CLEC14A, BMPR2, FLT1, SLCO2A1, EPAS1, GATA2) and lymphatic (PROX1, LYVE1, FLT4, and PDPN). Within vascular ECs, we found three main subtypes: arterial, capillary, and venous. Arterial ECs are exposed to higher pressure compared to capillary and venous ECs; as such, these cells exhibit more gap and tight junctions, extracellular matrix proteins, and protease inhibitors. These cells can be identified due to their higher expression of EFNB2, SOX17, BMX, SEMA3G, HEY1, LTBP4, FBLN5, GJA5, and GJA4. Capillary ECs facilitate gas exchange in the alveoli and include aerocytes and general capillary ECs (gCAPs) in an approximate one-to-two ratio. These terms were coined by Gillich et al. [48]. Capillary ECs were distinguished by high expression of CA4, PRX, RGCC, SPARC, and SGK1. Aerocytes express EDNRB, TBX2, FOXP2, CLEC4E, SPON2, PRKG, CHRM2, S100A4, EDA, HPGD, SOSTDC1 and do not express Weibel–Palade body-associated genes. gCAPs can be identified by the following genes: GPIHBP1, CD36, FCN3, BTNL9, BTNL8, CD14, IL7R, IL18R1. Finally, of the three vascular EC subtypes, venous ECs are exposed to the lowest pressure. Thus, these cells express genes that enable immune cell extravasation, including VCAM1, SELP, SELE, and ACKR1. Venous ECs are further subdivided into pulmonary venous (COL15A1neg), which are found in the lung parenchyma, and systemic venous ECs (COL15A1pos), which are localized to the airways and visceral pleura. In IPF, we observed loss in alveolar capillary cells corresponding to gCAPs and aerocytes [20] with a concomitant increase in venous ECs (COL15A1pos) in the lung parenchyma where they are never found. Immunhistochemistry (IHC) confirmed their presence in fibrotic and bronchiolized areas in IPF lungs paralleling the proximalization of the distal lung observed in epithelial cell populations [20].
Lung endothelial cells in COVID-19 also demonstrated distinct cell composition and phenotypic shifts compared to those of healthy controls. Delorey et al. identified several EC subsets in patients with COVID-19 [35]. These subsets included lymphatic (PROX1, MMRN1, RELN, PKHD1L1, CCL21, SEMA3D), arterial (VEGFA, EFNB2, DKK2, FBLN5, SERPINE2, CLDN10), venous (HDAC9, CPE, IL1R1, ACKR1, PTGS1), capillary aerocytes (EDNRB, HPGD, CA4, PRKG1, TBX2, RCSD1, EXPH5, EDA, FOXP2, CHRM2), two capillary subtypes (CA4, RUNX1, PRKCB and FCN3, VWF, PTPRB, NOSTRIN), and a mixed population (CD14, FCN3, GPIHBP1, SOSTDC1). Unlike Rendeiro et al.’s finding of a decreased proportion of endothelial cells in COVID-19 patients, the authors found that patients had significantly increased numbers of vascular and lymphatic endothelial cells (FDR < 0.001) [35]. These subtypes were also determined to have the highest significantly different gene expression profiles across all cell types in the lung, with 587 and 317 out of 1580 genes with increased expression in COVID-19 in lymphatic and vascular ECs, respectively. Though the authors do not explicitly report on changed pathways, Rendeiro et al. found that there was a significant enrichment in C5b-C9 complement activation and apoptosis pathways in differentially expressed genes in lung endothelial cells [34]. Higher proportions of venular and capillary endothelial ECs were also directly correlated with higher viral burden, possibly suggesting the role of these subtypes in disease [35]. Thus, endothelial cell changes in COVID-19 still present a muddled picture and require further investigation. Based on reported findings thus far, unlike changes in epithelial and myeloid cells, there was a limited similarity between fILD and COVID-19 lungs, potentially reflecting the distinct impact of viral pathogenesis on the endothelial compartment.
Mesenchymal compartmentPulmonary fibrosis is characterized by aberrant activation of fibroblast cells with accumulation of cells defined as myofibroblasts, changes in lung resident fibroblast populations, and excessive deposition of extracellular matrix proteins. In the last decade, a lot of studies investigated the molecular and metabolic changes involved in this differentiation. However, little is known about the diversity of fibroblast cellular populations in the IPF lung, mainly due to the lack of specific markers for the distinct populations. Single-cell analysis provides a more comprehensive view of cellular identity, where global transcriptomic features, rather than singular cell markers, are exploited. Single-cell analysis confirmed the relative increase of fibroblasts in general and of myofibroblasts in the pulmonary fibrosis patients compared to controls [20, 21]. These cells showed enrichment of fibrillar collagen contents of ACTA2 in the fibrotic lung versus normal lung. Despite a lot of effort dedicated to the investigation of the cellular source of myofibroblast, the origin of this cell is still debated. Habermann et al. identified four distinct lung fibroblast phenotypes expressing pathologic ECM proteins and localized in a different area of the fibrotic lung [21]. Myofibroblasts were predominantly localized in subepithelial regions around airways and areas of cystic remodeling, while HAS1hi fibroblasts appear restricted to the immediate subpleural region; PLIN2 + and other LUM + fibroblasts are found diffusely in parenchymal regions. The distinct phenotypes are probably not stable over time in vivo. Indeed, lung fibroblast populations demonstrate considerable plasticity. Application of lineage reconstruction technique partition-based graph abstraction suggested that pathologic, ACTA2-expressing IPF myofibroblasts are connected to a quiescent stromal population also present in control lungs [20]. Recent scRNA-seq data showing the higher ACTA2 expression in smooth muscle cells and pericytes suggest that α-SMA may not be a specific marker of pathologic fibroblasts producing the highest levels of extracellular matrix protein [
留言 (0)