
記住我
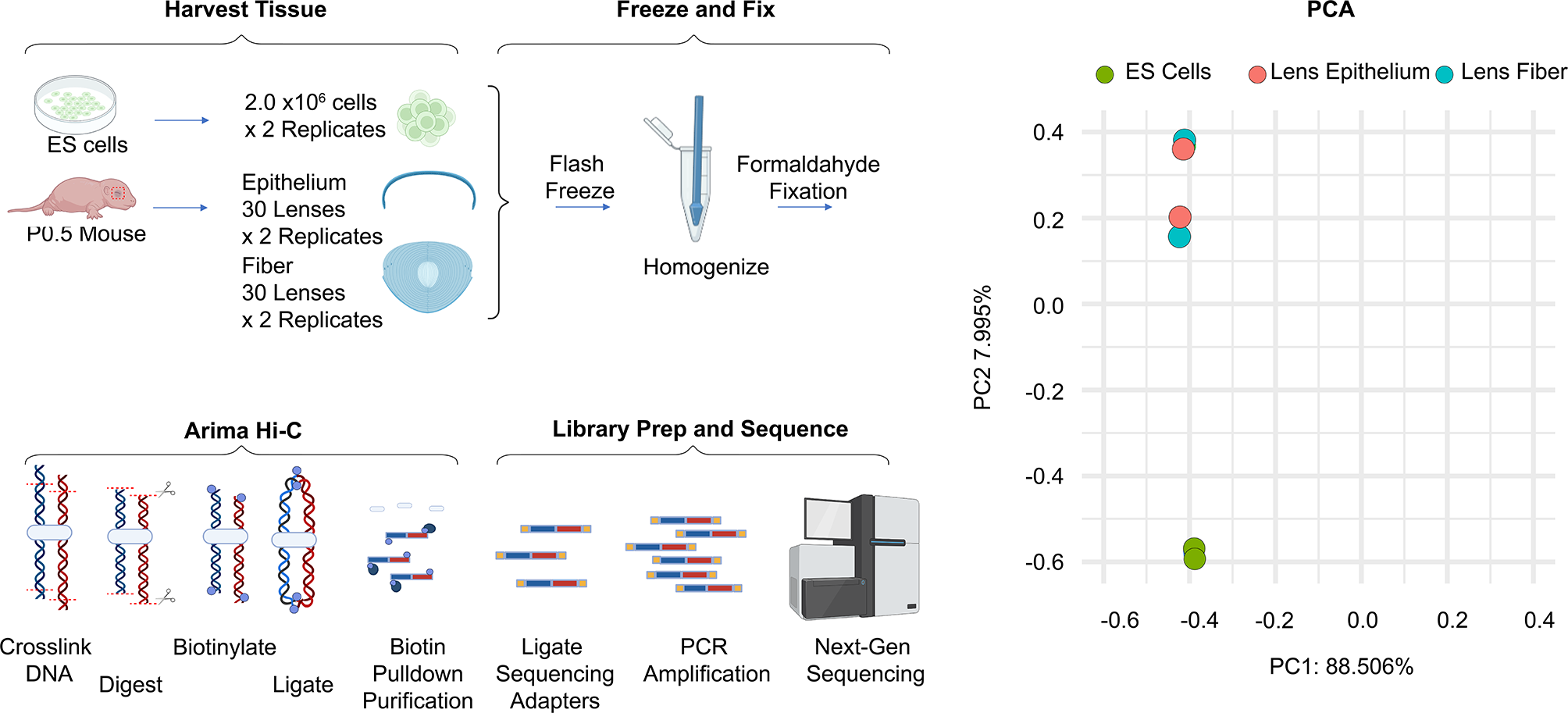


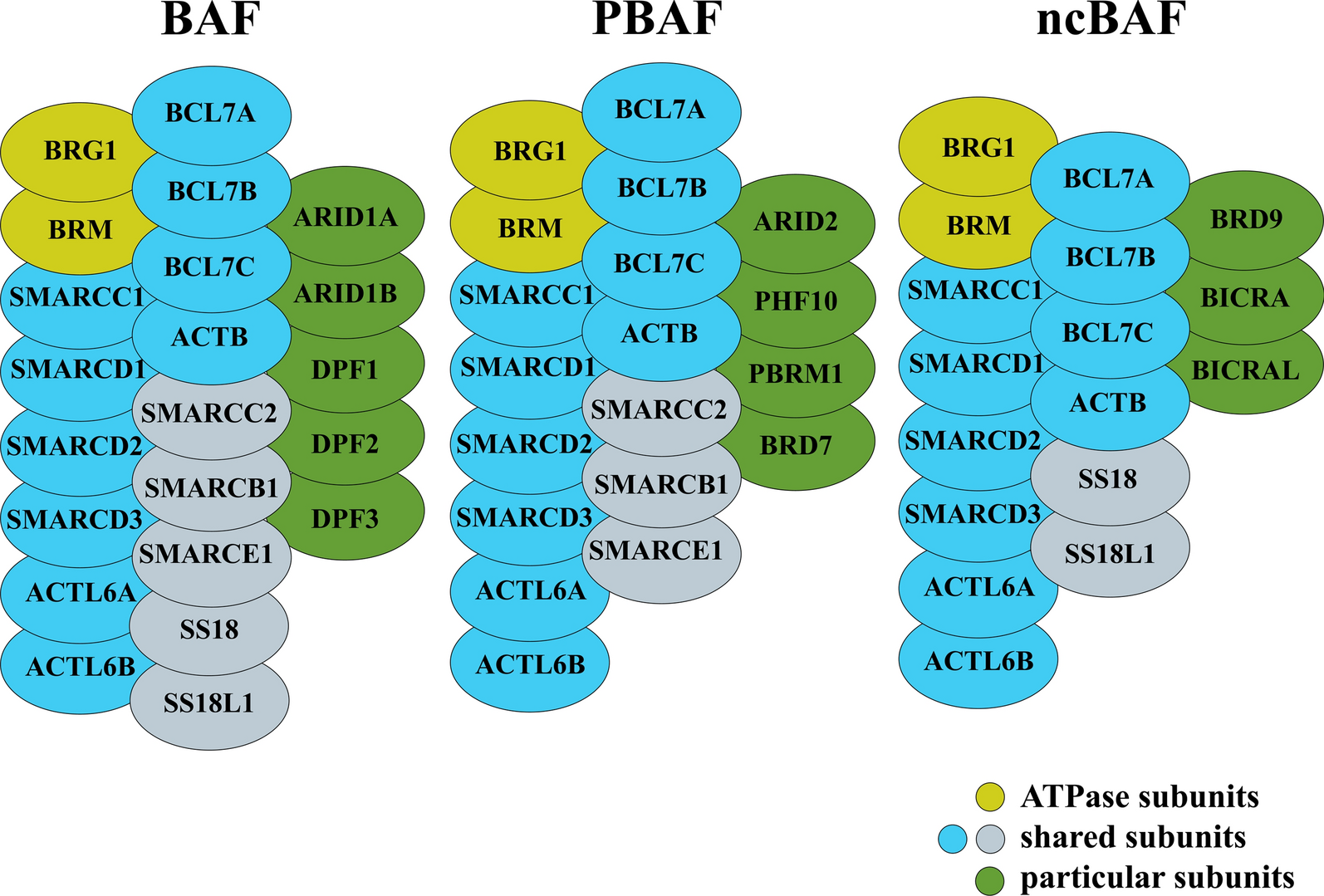
Eukaryotic nucleosomes, which package and regulate access to DNA, wrap 147 bp of DNA around an octameric core particle of two molecules each of the histones H2A, H2B, H3 and H4 [1]. Each of these histones has a histone fold domain (HFD) that consists of three α helices separated by two loops that together can interact with another HFD in an anti-parallel “handshake”. Despite sharing an HFD, each of the four core histones is distinct, with alignments between the different human core histones at best only on the edge of significance with E-values of 0.001 or higher. HFD proteins have a long history in all domains of life [2], but an innovation of eukaryotic histones is their ability to heterodimerize in specific pairs, H3 with H4 and H2A with H2B, that can further associate through four-helix bundles, H3 with H3’ and H4 with H2B, to form a central (H3–H4)2 tetramer flanked by two H2A–H2B dimers. In addition to the three-helix HFD, H3 has an additional αN helix that helps to wrap DNA, H2A has a short αN-helix and a C-terminal “docking domain” that helps to stabilize the interaction of the H2A–H2B dimers with the (H3–H4)2 tetramer, and H2B has an αC helix that together with its α3 helix forms the outer limit of the flat surface of the spool-like nucleosome. Each histone has an N-terminal and a C-terminal unstructured tail. The tails, especially the N-terminal tails of H3 and H4, have several conserved sites of post-translational modifications (PTMs) associated with gene activation or repression. A nucleosome may be further stabilized by the H1 ‘linker’ histone, which interacts with the DNA that links adjacent nucleosomes. H1 histones lack a HFD and have a separate origin from the other four histones [3], but are present in most eukaryotes [4]. A remarkable feature of the four core histones is that they are found in all eukaryotes [5, 6] spanning at least 1.6–2.4 billion years of the diversification of modern eukaryotes [7,8,9], and are among the most conserved proteins known. The mean amino acid identities in the HFDs to human core histones across 1208 eukaryotic genomes are 91%, 83%, 92%, and 93% for H2A, H2B, H3, and H4 [10]. Despite this strong conservation, some histones in certain protists such as H2B in Encephalitozoon cuniculi can have as little as 24% identity with human H2B yet are still able to form nucleosomes that bind DNA with little sequence preference [10]. In contrast to the general conservation of core histones, some histone variants, paralogs of the core histones, such as the centromere-specific H3 variant cenH3 and germline-restricted H2A variant H2A.B, have evolved rapidly and adapted for specialized functions [11, 12].
In the past two decades, homologs of the genes that encode these quintessentially eukaryotic proteins have been discovered in a growing number of double-stranded DNA virus genome sequencing projects, including in the giant viruses of the Nucleo-Cytoplasmic Large DNA Viruses (NCLDVs or Nucleocytoviricota). NCLDVs share genes from a core genetic content of ~ 50 genes that have been inferred to be present in a common ancestor of these viruses, though many genes have been lost or replaced in some lineages [13, 14]. Giant viruses have been variously defined by a genome size > 300 kb [15], a virion size > 200 nm [16, 17], or a proteome size of at least 500 proteins, sizes that compare to the sizes of bacteria, archaea, and picoeukaryotes [16, 18]. Their genes may encode components of the translation apparatus [19], enzymes of carbon metabolism [20], actin, myosin, and kinesins [21, 22], rhodopsins [23], and often a very large number of genes that lack homologs in other organisms (ORFans), challenging the “pickpocket paradigm” of viral genes as being derived largely or exclusively from their hosts [24], and raising the question of how often host genes are instead derived from viruses [25]. Some viruses, such as bracoviruses, have only one histone gene [26, 27], encoding a protein with high identity to the corresponding eukaryotic host histone, an apparent pickpocket’s prize from the host genome. In contrast, some NCLDVs have a complete set of all four histone genes or more, encoding highly divergent histones that appear to have anciently diverged from the corresponding eukaryotic histones prior to the diversification of modern eukaryotes [28,29,30], and which may be coupled in specific doublets [30,31,32] or even in triplets or quadruplets [20, 33]. Recently, cryo-EM structures of in vitro-assembled histone doublets from the viral family Marseilleviridae have been shown to form nucleosomes remarkably similar to eukaryotic nucleosomes [34, 35]. How did these histones come to be encoded in viral genomes? Do these proteins form nucleosomes in vivo? Do they interact with the host genome, or with the viral genome, or both, or neither? What are their functions? Can they be post-translationally modified like eukaryotic histones?
In this review, we collate data on the occurrence of viral histones, most of which have not been investigated, and summarize what is known about the rapidly developing field of viral histones. We discuss their possible functions, how the viral life cycle may influence their properties, whether viral replication occurs in the nucleus or cytoplasm, and discuss scenarios of their origins and evolution, particularly in the context the viral karyogenesis hypothesis of nuclear origin. Our goal is to draw attention to the perplexing diversity of viral histones and spark further investigations into their roles in viral and cellular evolution.
Viruses that use eukaryotic histonesHistones famously package and protect DNA in eukaryotes and also have regulatory roles for processes that access DNA, such as transcription and replication. Viruses have evolved a variety of strategies to package their DNA into capsids without using histones (reviewed in Ref. [36, 37]), including even the giant mimivirus, which uses glucose-methanol-choline oxidoreductases to enclose its genome in a helical protein shell [38]. Some dsDNA viruses that do not encode histones can nevertheless utilize eukaryotic histones for their own packaging, regulation, or protection, and provide a point of reference in considering viral-encoded histones. During lytic infection of human foreskin fibroblasts, unchromatinized herpes simplex virus initially acquires histone H3-containing nucleosomes with the heterochromatic silencing PTMs H3K9me3 and H3K27me3 upon entry into the nucleus, in a defensive effort of the host cells to silence the virus. Later the viral immediate-early protein ICP0 reduces H3K9me3 on viral DNA and the immediate-early protein VP16 recruits histone acetyltransferases for H3 acetylation, a PTM associated with active chromatin [39, 40]. During latent infection in sensory neurons, in contrast, the herpesvirus proteins help to promote silencing of the episomal genome with the PTMs H3K9me3 and H3K27me3 [41, 42]. The silenced genome resides permanently in the neuron, but can occasionally re-activate to produce infectious virions.
In contrast to herpesvirus, the papillomavirus genome is packaged with host histones in the virion [43]. These histones are enriched for PTMs associated with active chromatin, which presumably reflects the chromatin state late in infection when genomes are loaded into the virions, but which may also serve to promote early transcription and replication of the viral genome upon new infection, as well as to help the viral genome evade detection by host DNA-sensing mechanisms [44]. The nucleosomes in the virion are also enriched for the replication-independent (RI) H3 variant H3.3, which in cells replaces the replication-coupled (RC) variants H3.1 and H3.2 at active sites of nucleosome turnover and is likely the major H3 variant available for packaging the papillomavirus genome during infection of non-replicating differentiated cells. These viruses demonstrate the ability to adapt to and take advantage of a eukaryotic chromatin environment for epigenetic gene regulation and virion packaging, roles that can be expanded in viruses that encode their own histones.
Single viral histonesA number of viruses encode a single histone that is highly similar to its eukaryotic counterpart and likely functions by being incorporated into host nucleosomes. The H4 gene of bracoviruses is the best-studied of these, but single H2Bs and H3s are also known.
Bracovirus H4 (CvBV-H4)Polydnaviruses have been considered to be endosymbiont proviruses in the genomes of certain subfamilies of ichneumonid and braconid wasps, which are themselves endoparasitoids on insect larvae, especially lepidopteran larvae [45]. Female wasps of the microgastroid complex of braconid subfamilies, encompassing tens of thousands of species, lay eggs in lepidopteran host larvae and simultaneously inject virion particles (Fig. 1). Virion packaging genes are only transcribed in the calyx cells of the pupal-to-adult female ovary, where they package DNA circles amplified from the proviral segments that encode numerous virulence genes [46]. The DNA circles in the virions do not encode viral replication genes. Instead the viral genome is endogenized in the wasp genome and passed on vertically from wasp to wasp. Because the virions do not encapsidate the information for their own replication, a recent definition considers that bracoviruses and ichnoviruses are not truly viruses, but “polydnaviriformids”, though they clearly descend from viruses [47]. The viral genes in bracoviruses are derived from a beta nudivirus that integrated into an ancestral wasp some 100 million years ago [48], whereas different viruses, including an unknown member of the NCLDVs, gave rise to the ichnoviruses [49, 50].
Fig. 1
Life cycle of Cotesia and bracovirus. Female Cotesia wasps lay eggs in larvae of moths such as Plutella xylostella and also inject bracovirus virion particles carrying DNA circles that integrate into the chromosomes of the parasitized larvae and favor development of the wasp larvae over the moth larvae. The bracovirus provirus is resident in the wasp genome and transmitted directly to offspring
The DNA circles in the virions integrate into the chromosomes in host cells, where expression of the virulence genes interferes with the host immune response and development, favoring the growth of the wasp larvae at the expense of the host, which is usually killed [45]. The virulence genes may be derived from the wasp, from transposable elements, or from unknown sources. One of the virulence genes in the wasp Cotesia vestalis (synonym C. plutellae), which parasitizes the larvae of the diamondback moth Plutella xylostella, is a viral-encoded histone H4 [26, 27]. The bracoviral H4 (CvBV-H4; accession numbers for all histones discussed in this review are found in Additional file 1: Table S1) is encoded on one of 35 genomic segments that make up the bracovirus genome of 351 kb [51, 52]. CvBV-H4 is ~ 87% identical to the H4 of P. xylostella and other insect H4s in the HFD and has conserved lysines corresponding to PTM sites K5, K8, K12, K16, and K20 of the eukaryotic H4 tail (Additional file 2: Fig. S1), but has an additional 38 amino acids at its N-terminus, including nine additional lysines [27]. CvBV-H4 is expressed in the nuclei of hemocytes of parasitized P. xylostella, which show greater H4 acetylation than unparasitized hemocytes, suggesting CvBV-H4 is acetylated. In eukaryotes, acetylation of H4 is associated with gene activity, while trimethylation of H4K20 is strongly associated with gene silencing [53]. CvBV-H4 is recovered in bulk nucleosomes from parasitized P. xylostella together with the endogenous H4 and other histones [54]. In vitro CvBV-H4 forms octamers with all four core histones, but not when H4 is omitted, suggesting that the octamers contain two H2As, two H2Bs, two H3s, and one each of CvBV-H4 and H4 [55]. CvBV-H4 suppresses the host immune response and delays P. xylostella larval development, favoring the growth of C. vestalis, dependent on its N-terminal tail [54, 56,57,58]. Chromatin immunoprecipitation of P. xylostella genomic sites enriched in CvBV-H4 revealed 51 sites in common between parasitized P. xylostella and non-parasitized P. xylostella transiently expressing CvBV-H4 that were not enriched in non-parasitized larvae or larvae expressing a truncated CvBV-H4 gene that lacks the N-terminal tail. Genes within 1 kb of these sites have roles in development, metabolism, immunity, signaling, and gene expression [59]. Given that H4 is deposited in insect chromatin as an H3–H4 dimer either with RC assembly of H3.2 by CAF1 or with RI assembly of H3.3 by HIRA [60], it seems likely that these 51 sites are subject to high nucleosome turnover and that CvBV-H4 is primarily deposited with H3.3 by HIRA in differentiated hemocytes and other cells. A role for the viral H4 tail in stabilizing the viral histone at specific sites is suggested by the enrichment of transiently expressed CvBV-H4, but not its truncated derivative, at these sites. Expression of CvBV-H4 results in 81 up-regulated genes, half of unknown function, and 221 down-regulated genes, 70% of which were predicted to have functions in development and metabolism [61]. Among the down-regulated genes are H4 [54], a lysine demethylase, and a SWI/SNF chromatin remodeler [62].
Virulence genes are expected to help Cotesia species adapt to different hosts and may therefore show signatures of positive selection. Comparison of CvBV-H4 with the viral H4s of C. congregata and of two incipient species, C. sesamiae kitale and C. sesamiae Mombasa, detected positive selection both in the tail and in the HFD [52]. Further examination of viral histones in 17 C. sesamiae populations found that polymorphisms that might reflect adaptation were primarily indels of seven amino acids in the tail (Additional file 2: Fig. S1).
The presence of bracovirus H4 genes in all investigated Cotesia species and their high sequence identity with insect H4s strongly suggests that an endogenous H4 gene in the ancestor of Cotesia species some 17 million years ago [63] was recruited to the bracovirus virulence genes to become the ancestral viral H4. The conservation of post-translational modification sites and incorporation of CvBV-H4 into host nucleosomes indicates that bracovirus H4s have been constrained to interact with the nucleosomes of the lepidopteran host species, and finding positive selection suggests that this interaction is subject to change as populations diverge. The extended tail of CvBV-H4 that suppresses the host immune response and delays larval development appears to have been weaponized by parasitic Cotesia species to gain an advantage against their hosts. The use of histones as weapons in microbial warfare has ample precedent [64].
Metagenomic viral H4While bracoviruses may have recently acquired H4 from their obligate mutualistic symbionts, more ephemeral symbiotic interactions may also lead to viral acquisition of histones. Co-infection of Acanthamoeba polyphaga by Marseillevirus and bacterial symbionts led to the proposal that amoebae can serve as “melting pots” for horizontal gene transfers between viruses, bacteria and their hosts [17]. Transfers can occur from host to virus or virus to host, with the latter occurring at about half the frequency of the former [25]. A marine metagenome-assembled genome (MAG) related to pandoraviruses (ERX552244.21) encodes an H4-like protein [20] that is 77% identical to human H4, and is surprisingly 93% identical to an H4-like protein encoded in a MAG [65] attributed to a Verrumicrobiales bacterium (MAD25601.1). Bacterial and archaeal MAGs sometimes contain contaminating sequences from NCLDVs. The viral H4, the bacterial H4, or both could be contaminants, but they may also potentially be recent horizontal gene transfers from the same or similar eukaryotic hosts. With only a few changed residues in the N-terminal tail and HFD and all tail lysines conserved, the H4-like protein of ERX552244.21 is likely to be incorporated into nucleosomes of its unknown host.
Pandoraviridae H2BsThe pandoraviruses have linear genomes up to 2.5 Mb [66] and are the largest of the giant viruses. Pandoravirus virions are engulfed into Acanthamoeba castellanii cells by phagosomes, which fuse with lysosome-like organelles that seem to stimulate the uncoating of the virions [66,67,68]. The capsid opens and the internal membrane fuses with the host phagosome in which they are engulfed, spilling their genome into the cytoplasm. A viral factory assembles in the cytoplasm that can recruit mitochondria and membranes, eventually leading to virion assembly and release by either exocytosis or lysis [68]. About 10% of pandoravirus genes with homologs in eukaryotes have introns, which, along with the absence of transcriptional machinery in the virion, strongly suggests that at least some viral transcription takes place in the nucleus, which disintegrates late in the infection [66]. About two thirds of genes in pandoraviruses are ORFans unique to pandoraviruses [67].
Among the minority that have eukaryotic homologs is H2B, which is present in eight completely assembled pandoravirus genomes, although homologs of H2A, H3, and H4 are absent. Pandoravirus H2Bs are 70%-75% identical to H2B from A. castellanii or 64–73% identical to human H2B in the HFD and in the αC helix, retaining the K120 ubiquitylation site (as numbered in human H2B), but they have modestly extended C-terminal tails, and the long N-terminal tails are divergent from eukaryotic H2Bs and from each other (Additional file 2: Fig. S2). They have ~ 15 potentially modifiable lysines in the N-terminal tail, which are similar in number though not in exact positions to the lysines in A.castellanii or human H2B tails. The pandoravirus H2B N-terminal tails contain 8–10 acidic residues in the first 50 amino acids while eukaryotic H2Bs lack this acidic region.
Though most pandoravirus H2Bs are ~ 200 amino acids in length, the predicted H2B protein from Pandoravirus inopinatum is truncated by a frameshift. In contrast, the tail of P. celtis has an additional 121 amino acids at the amino terminus for a total of 319 amino acids (Additional file 2: Fig. S2). A model of new ORFan gene formation in pandoraviruses proposes that new genes arise from intergenic regions that acquire transcription and translation initiation and termination signals and are then selected for function [69]. The additional 121 amino acids in the tail of P. celtis are consistent with such a model. Comparison of the corresponding intergenic region in the sibling virus P. quercus, which encodes an H2B of 198 amino acids, with the region encoding the extra 121 amino acids in P. celtis, reveals that the region 5′ to the P. quercus H2B gene has undergone a two base pair duplication in P. celtis. This causes a frameshift, which together with 10 single base pair substitutions including the conversion of a stop codon to a tryptophan allow an upstream methionine codon to initiate the extended P. celtis tail. The H2B initiation codon in P. quercus is still present in P. celtis, and it is possible that translation initiation 121 codons upstream is a misprediction, but the loss of a stop codon that permits upstream initiation may indicate that the extended N-terminus has been selected for some advantage.
The function of pandoravirus H2B is unknown. The protein is not present in the virion proteomes of five pandoraviruses [67, 70] suggesting the protein functions only in the host cell. Indeed, histone transcripts from pandoravirus relatives have been reported from virocells (infected cells reprogrammed by a virus) from the marine microbial community of the California Current [22]. The high amino acid identity of pandoravirus H2Bs with eukaryotic H2Bs in the HFD and absence of other viral-encoded histones strongly suggests that the protein assembles with host histones into nucleosomes in the nucleus, similar to CvBV-H4. Like CvBV-H4, pandoravirus H2B might suppress or re-direct the host genome to favor viral replication. The lysines in the tail may be acetylated and conservation of H2BK120 suggests that pandoravirus H2Bs can be ubiquitylated co-transcriptionally. H2BK120ub1 facilitates methylation of H3K4, H3K36, and H3K79, all of which are associated with active transcription [53]. The high identity with eukaryotic H2Bs in the HFD also suggests that this gene was acquired by an ancestral pandoravirus from a eukaryotic host and maintained under strong selection even as the pandoraviruses have diversified their genomes with new genes to the extent that the core genome common to six species represents only 15–29% of each individual genome [70].
Single H3 histonesSeveral viruses encode an H3-like histone but not other histones, suggesting that these viral histones also interact with host histones to have their effects. Consistent with this, they generally have high similarity to eukaryotic H3s in the HFD (Additional file 1: Table S1, Fig. S3). Since the chaperone recognition site on α2 of H3 determines whether H3/H4 dimers can assemble into nucleosomes by the RC pathway in replicating cells and/or the RI pathway in non-replicating cells, this site is likely to be critical to the ability of the virus to interact with the host chromatin. Some but not all of these viral histones have longer N-terminal tails, similar to CvBV-H4 and pandoravirus H2Bs.
Manila clam xenomavirus is a recently described virus that may be related to papillomaviruses, polyomaviruses, and adomaviruses, which are small tumor viruses that are thought to have derived from circular Rep-encoding single-stranded DNA (CRESS) viruses [71]. Xenomavirus encodes a histone H3-like protein that is 77% identical to the H3.2 or H3.3 of Manila clam (Ruditapes philippinarum) in the histone fold domain and the αN helix. In the α2 helix, where the sequences SAVL (H3.2) and AAIG (H3.3) determine RC or RI assembly, respectively, xenomavirus H3 has SAIL, which would likely be permissive for assembly by either pathway if the viral N-terminal tail can substitute for the H3.2 tail, which is required for RC assembly [
留言 (0)