
記住我


Toxic and allergic epitopes should be avoided since they can endanger the goal of vaccine development. Antigenicity, allergenicity, toxicity, and immunogenicity of anticipated epitopes were assessed using Vaxigen v2.0, AllerTOP v2.0, Toxinpred, and the IEDB class I immunogenicity tool, respectively. For further investigation, epitopes with positive immunogenicity scores, high antigenicity, non-allergenicity, and nontoxic nature were chosen.
Prediction of linear B-cell epitopesThe ABCpred server was used to examine the protein's amino acid sequences for the presence of linear epitopes. A total of 30 epitopes of length 10 mer were predicted as mentioned in Additional file 1: Table S1. Only two epitopes (DADGVEKKVL and DIKLTIDAKA) were chosen for the final vaccine construct based on the above-mentioned criteria (Table 1).
Table 1 Final selected B-cell epitopes from Enterococcus faecium penicillin binding protein and their corresponding immunogenic propertiesPrediction of cytotoxic T lymphocytes epitopeMHC Class I binding peptides were predicted using the NetMHCpan 4.1 server (Additional file 2: Table S2). The top five peptides from having high antigenicity and positive immunogenicity score were shortlisted for the final vaccine construct and are shown in Table 2.
Table 2 Predicted CTL epitopes from Enterococcus faecium penicillin binding protein to design multi-epitope vaccine construct with their corresponding MHC Class I alleles and their immunogenic propertiesPrediction of helper T lymphocytes epitopePeptides that bind to MHC class II molecules were predicted using the IEDB server's MHC class II binding peptide prediction tool. The IEDB-recommended approach was employed for MHC class II binding, and a reference set of 27 alleles was used. As shown in Additional file 3: Table S3, a total of 10 HTL epitopes and their matching HLA class II binding alleles were predicted for the protein sequence. Based on the above criteria, the top two HTL epitopes (DKFIFGEDLDLPISM and DSLGGKAGSTVATTP) were selected for the final vaccine construction (Table 3).
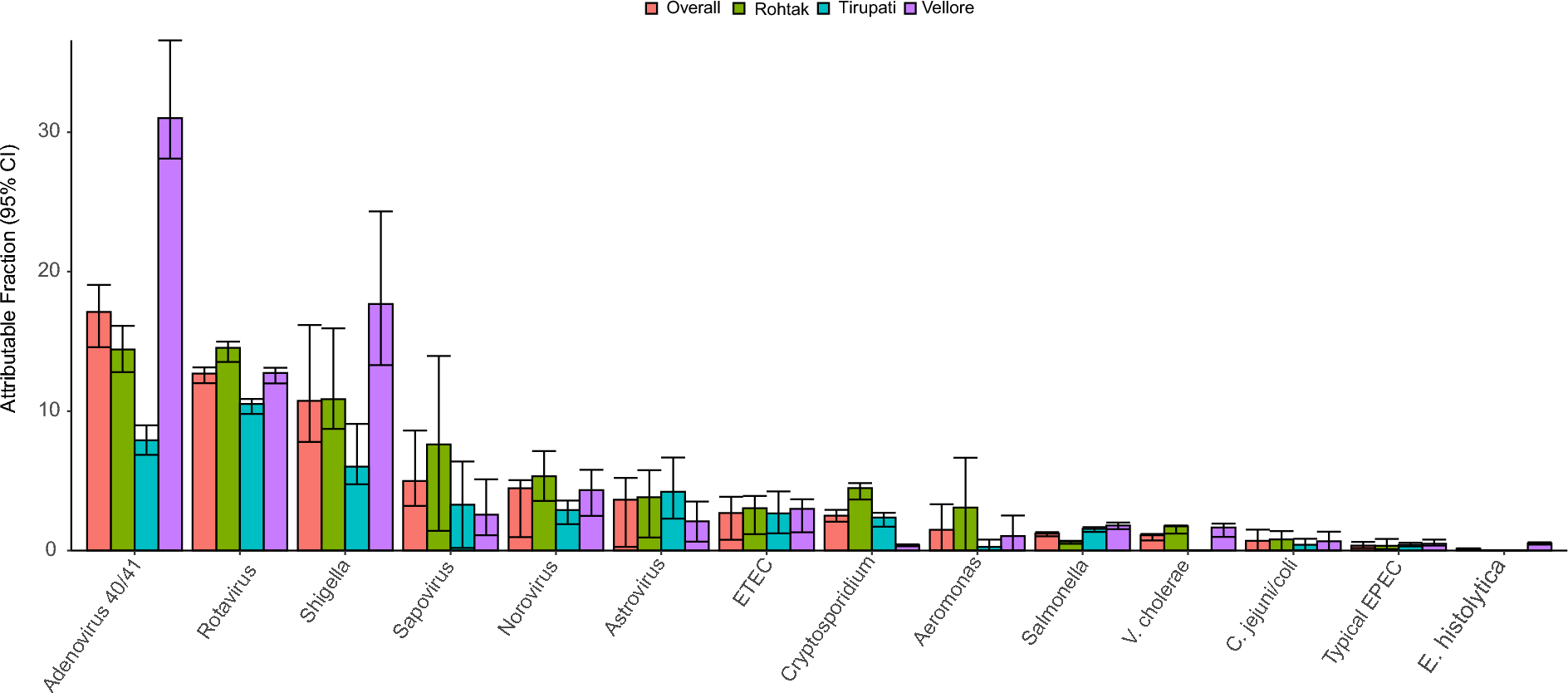
Table 3 Predicted HTL epitopes from Enterococcus faecium penicillin binding protein to design multi-epitope vaccine construct with their corresponding MHC Class II alleles and their immunogenic propertiesPopulation coverage analysis of the selected epitopesIndividual and combinatory population coverage for the seven chosen T-cell epitopes (5 CTL and 2 HTL) with their binding HLA alleles was calculated, as shown in Additional file 4: Table S4. Global population coverage of 99.25% and 96.20% individuals will respond to the selected epitopes in India. West Africa had the highest population coverage of 99.93%.
Construction of the multitope vaccine candidateFinally, a 172 amino acid long multi-peptide vaccine was created by connecting several epitopes such as LBL, CTL, HTL with suitable linkers at appropriate places. The final subunit vaccine was constructed using three linkers: KK, AAY, and GPGPG. The AAY and GPGPG linkers were introduced to the intra-epitope site to connect the CTL and HTL epitopes, respectively. Then, to boost the effectiveness of the immune response, human beta-defensins (accession id: P81534) of 67 amino acid length was added to the N-terminal end of the designed vaccine sequence as a molecular adjuvant. Figure 2 depicts the arrangement of adjuvant and epitopes with their connecting linkers.
Fig. 2
The structural arrangement of B and T-cell epitopes along with linkers and adjuvant for the final multi-epitope vaccine construct
Physicochemical features, solubility, and secondary structure predictionThe aliphatic index, GRAVY, Theoretical PI, instability index, and molecular mass of the multi-epitope vaccine were calculated to be 73.90, −0.472, 9.38, 18.85, and 18,294.07 Da, respectively, based on the ProtParam server data. The assessment of the protein's molecular weight was the most important aspect throughout this phase [34, 35]. Proteins with a molecular weight of < 110 kDa are thought to be more suitable since they can be easily isolated and effectively put to vaccine development [36, 37]. Based on the study of physicochemical qualities, the vaccine design was anticipated to be stable, basic, hydrophilic, thermostable, and highly soluble following over-expression in E. coli. Furthermore, the vaccine's half-life was determined to be 30 h in human reticulocytes (in vitro), over 20 h in yeast (in vivo), and over 10 h in E. coli (in vivo). Additional file 5: Table S5 summarises the physicochemical parameter analysis results.
According to the results of the PSIPRED server, random coils are formed by 99 amino acids, α-helix by 26 amino acids, and β-strands are formed only by 47 amino acids. The overall secondary structure prediction of the developed vaccine results concluded that 57.55% are random coils, 15.11% form α-helix and 27.32% are β-strands as represented in Fig. 3.
Fig. 3
Secondary structure prediction of the final multi-epitope vaccine construct by using PSIPRED tool
Tertiary structure prediction and validation of the vaccine constructThe Robetta server was used to predict five tertiary structures for the vaccine construction, which were then analyzed using a Ramachandran plot to choose the optimum model. Model 3 was chosen because it provided the best arrangement of residues in allowed regions (Fig. 4). The Ramachandran plot showed that most of the residues were found in the favored (81.4%), allowed (12.9%) regions, and 2.1% in generously allowed regions, while 3.6% residue was present in the disallowed region. The model's ERRAT quality factor and Verify-3D score were 92.661 and 95.93; respectively indicate that this structure is energetically stable. The Ramachandran plot, ERRAT, and verify3D of the multi-epitope peptide structure are shown in Fig. 5.
Fig. 4
Homology modeling of the three-dimensional structure of the final multi-epitope vaccine construct
Fig. 5
Several structure validations tools results confirmed the modeled multi-epitope vaccine structure to be reliable and accurate
Disulfide engineering for vaccine protein stabilityAfter running the vaccine sequence through the Disulfide by Design 2.13 tool, a total of 13 possible residue pairings that potentially form a disulfide bond were found, as shown in Additional file 6: Table S6. Taking the bond energy and χ3 parameters into consideration, only one pair of residues was chosen because their scores fit conventional standards, i.e. the bond energy should be less than 2.2 kcal/mol and the χ3 angle should be between −87° and + 97°. As illustrated in Fig. 6, a mutation pair was created on the residue pair CYS11–CYS18, which had bond energy of 1.14 kcal/mol and a χ3 angle of + 96.08°.
Fig. 6
Disulphide engineering of the vaccine protein. Residue pairs showed in purple (CYS11) and olive (CYS18) spheres were mutated to Cysteine residues to form disulphide bridge between them
Mapping of discontinuous B-cell epitopes in the vaccine proteinB lymphocytes boost humoral immunity by secreting antibodies and cytokines that neutralize foreign antigens [38, 39] That's why it is essential to have a sufficient number of B-cell epitopes inside the protein domain. The existence of conformational B-cell epitopes was analyzed using the ElliPro tool of the IEBD server, which found 4 conformational B-cell epitopes of 14–39 residues (Additional file 7: Table S7) with scores ranging from 0.596–0.809, as shown in Fig. 7.
Fig. 7
The conformational B-lymphocyte epitopes present in the vaccine. The yellow spheres showing epitopes containing (A) 20 residues (AA 153–172) with 0.809; (B) 14 residues (AA 69, AA 71, and AA 88–99) with 0.745; (C) 39 residues (AA 11, AA 13–16, AA 24–40, AA 42, and AA 44–59) with 0.621; (D) 27 residues (AA 1–2, and AA 127–151) with 0.596
Molecular docking of vaccine with TLR4 receptorTo assess the binding affinity between the vaccine construct and TLR4, docking analysis of vaccine protein with TLR4 was undertaken. Molecular docking was performed by using the grid box set in the active site region. The lowest binding energy was –856.8, and the center energy between the ligand and receptor was −671.1. Pymol was used to visualize the interactions between the TLR4 receptor and the vaccine candidate, and the docked complex is shown in Fig. 8. Residues of the vaccine construct and TLR4 receptor were found to make polar contacts are LYS43-ASN4, ASP41-ASN4, GLU88-ARG17, GLU88-LYS8, ARG86-LYS8, SER85-LYS131, ASP83-LYS131, GLU86-GLY126, GLU86-ARG125, GLU134-GLY130, ARG233-GLY130, ARG233-GLY130, ARG233-PRO136, GLU265-LYS62, TYR183-LYS61, TYR183-ARG12, ASN159-ARG12, TYR185-LEU60, GLN187-LYS57, and ASP215-LYS57 with the distance of 1.7 Å, 1.9 Å, 1.8 Å, 1.8 Å, 2.4 Å, 1.8 Å, 1.7 Å, 1.8 Å, 1.7 Å, 2.3 Å, 1.7 Å, 1.8 Å, 1.7 Å, 2.2 Å, 1.8 Å, 1.7 Å, 1.6 Å, 1.9 Å, 1.7 Å and 1.8 Å, respectively.
Fig. 8
Molecular interaction of multi-epitope vaccine construct docked with TLR2
Molecular dynamic (MD) simulationMD simulation in an aqueous environment was used to assess the stability of the protein–protein complex. The simulation time was displayed against the produced vaccine-TLR4 complex. according to the results, the cubic box was successfully created under intermittent boundary circumstances, and neutralization and energy reduction were carried out efficiently. Here we have run a simulation for 100nsffa. The produced vaccine's RMSD plot (as a ligand) indicated values in the 0.1–0.2 nm range. Result of the root mean square deviation has been shown in Fig. 9. It is noteworthy that the system was equilibrated in a shorter period. The RMSF plot shows that the complex is overlapped except at the N and C terminal of the protein as the substrate is interacting in this region (Fig. 9). The Substrate interacting region of the system is more fluctuating.
Fig. 9
Root mean square deviation (RMSD) and root mean square fluctuation (RMSF) analysis of protein backbone and side chain residues of MD simulated vaccine construct
The hydrogen bond is vital in preserving protein secondary structure, therefore an accurate description of hydrogen bond interaction is critical in protein folding simulation. Hydrogen bonding is an important nonbonded interaction in system simulation, which is dominated by electrostatic interaction. The number of hydrogen bonds is calculated using a sigmoidal function of the donor–acceptor distance as a function of hydrogen bond strength. The peak in Fig. 9 is quite high, demonstrating that integrating the polarisation effect strengthens hydrogen bonding. Furthermore, any modification in protein motif and shape can be evaluated by Gyration Radius (Fig. 9). The spherical state information about total protein volume distribution is obtained by this parameter. The results suggest that the Gyration Radius of the complex is more downwards that is a clue of constructed secondary structures and it seems that this is a result of the limitation of the main chain movement of the system.
Codon adaptation and in silico cloningSince differences in codon use result in limited translation of foreign genes, codon adaptation is the greatest strategy to improve translational efficiency. The JCat tool was utilized to improve our developed vaccine's codon use in relation to the E. coli K12 strain. The optimized sequence had a GC content of 51.93%, indicating effective expression in the E. coli host with a Codon adaptation index (CAI) of 1. The results showed that following adaptation, the prokaryotic ribosome binding site, restriction enzyme cleavable sites and rho independent transcription terminators were all eliminated. Following that, the vaccine construct's modified codon sequence was introduced into the E. coli expression vector PET28a (+) between the EcoRI and BamHI restriction sites, as shown in Fig. 10. A 6-histidine tag was also inserted to facilitate immune-chromatographic purification of the recombinant vaccine. As a result, the clone's length was 5889 bp.
Fig. 10
Restriction cloning of final multi-epitope vaccine by using pET28a (+) expression vector in the in silico space. Black circle indicates the vector, and the magenta part is the place where the vaccine is inserted
留言 (0)