Cell culture
SW480 (human colorectal carcinoma overexpressing EGFR) was kindly gifted by Dr. Michael Jakupec (Department of Inorganic Chemistry, University of Vienna, Austria). The cells were cultured as adherent monolayers in plastic tissue culture dishes in complete growth medium Eagle's Minimum Essential Medium (EMEM) (ATCC, USA) supplemented with 10% (v/v) heat-inactivated calf bovine serum (ATCC, USA) and 100 U/mL antibiotics (Sigma-Aldrich, USA), 0.1 mM non-essential amino acid and 1 mM sodium pyruvate. NIH/3T3 (EGFR-negative mouse fibroblast) was kindly gifted by Dr. Chomdao Sinthuvanich (Department of Biochemistry, Faculty of Science, Kasetsart University, Thailand). The cells were cultured in DMEM (Gibco, Carlsbad, CA, USA) containing 10% fetal bovine serum (ATCC, USA) and 100 U/mL antibiotics (Sigma-Aldrich, USA). Cells were maintained at 37ºC in a humidified incubator in an atmosphere with 5% CO2 and replenished every 3 days before the experiment set up. The cells were maintained at 37ºC in a humidified incubator in an atmosphere with 5% CO2.
Cell viability assay
The R9VH36-nanobody was dissolved in PBS at the concentration of 100 µM. Gefitinib (Santa Cruz Biotechnology, USA) was dissolved in DMSO (Loba Co., India) at the concentration of 10 mM. The final concentration was obtained by dilution in a complete culture medium. The cancer cell lines viability assay was performed using MTT assay (PanReac AppliChem, Germany) according to the manufacturer’s instructions. SW480 and NIH/3T3 cells were seeded on 96-well microtiter plates (5 × 105 cells/mL) in 100 μL of complete growth medium and allowed to attach overnight at 37 °C, 5% CO2. The next day, the cultured cells were treated with varying concentrations of R9VH36 (two-fold dilutions, starting at 10 µM) and gefitinib (two-fold dilutions, starting at 100 µM) for 72 h, 100 μL/well MTT reagent (0.5 mg/mL) was added to each well and incubated for an additional 4 h at 37ºC. The medium containing non-metabolized MTT was then aspirated, and 50 μL DMSO was added to solubilize the reduced formazan product. Finally, the absorption was then measured at excitation and emission wavelengths of 550 and 590 nm, respectively by a microplate reader (BioTek Synergy HTX, U.S.A.). Based on spectrophotometric measurements, the cell viability was calculated compared to the control cells (the absorbance of the control cells as 100% viability). The 50% inhibitory concentration (IC50) value was calculated from the graph of the dose–response by the following equation,
$$y=Bottom+\left(\frac^}\right)$$
where Bottom is the minimum values, Top is the maximum values, y is the cell index, and X is the testing concentration in logarithm unit.
Three independent experiments were performed with triplicates-treatment preparation. The IC50 of testing samples was used in the proteomics analysis.
Sample preparation for proteomics analysis.
The cells were placed in a T25 flask (Nest, U.S.A.) (~ 107 cells/flask) in triplicates. The day after, the cells were then replaced with a fresh medium containing 10 nM of R9VH36 or gefitinib for 1 h. The treated cell lysates were lysed in lysis buffer (10 mM HEPES/NaOH, pH 7.4, 0.25 M sucrose, 10 mM NaCl, 3 mM MgCl2, 0.5% Triton X-100) supplemented with a protease inhibitor cocktail (Sigma-Aldrich Co., USA). The supernatant was collected by centrifugation at 12000 g. and subsequent to ice-cold acetone precipitation. After precipitation, all samples were reconstituted in sample buffer (6 M Urea, 2 M Thiourea, 0.05% SDS, 10 mM NaCl). The protein solution was diluted in 10 mM ammonium bicarbonate at 1:20 ratio (v/v). The total protein (25 µg) was subjected to gel-free based digestion. Next, sulfhydryl bond reduction was performed using 5 mM DTT (Sigma Aldrich Co.) in 10 mM ammonium bicarbonate at 25 °C for 3 h and sulfhydryl alkylation using IAA (Sigma Aldrich Co.) at room temperature for 30 min in the dark. All samples were enzymatically digested for 16 h. The tryptic peptides were cleaned-up using C18 Zip-tip (Merck Millipore, USA) and reconstituted in 0.1% formic acid before being subjected to LC–MS/MS.
LC–MS/MS setting for tryptic peptide analysis
The tryptic peptides were analyzed using tandem mass spectrometers, Orbitrap HF hybrid mass spectrometer combined with an UltiMate 3000 LC system (Thermo Fisher, USA). The tryptic peptides were first desalted on the line of a reverse-phase C18 PepMap 100 trapping column, before being resolved onto a C18 PepMapTM 100 capillary column with a 135-min gradient of CH3CN, 0.1% formic acid, at a flow rate of 300 nL/min. Peptides were analyzed by applying a data-dependent top10 method consisting of a scan cycle initiated by a full scan of peptide ions, followed by high-energy collisional dissociation and MS/MS scans on the 10 most abundant precursor ions. Full scan mass spectra were acquired from m/z 400 to 1600 with an AGC target set at 3 × 106 ions and a resolution of 70,000. MS/MS scan was initiated when the ACG target reached 105 ions. Ion selection was performed applying a dynamic exclusion window of 15 s.
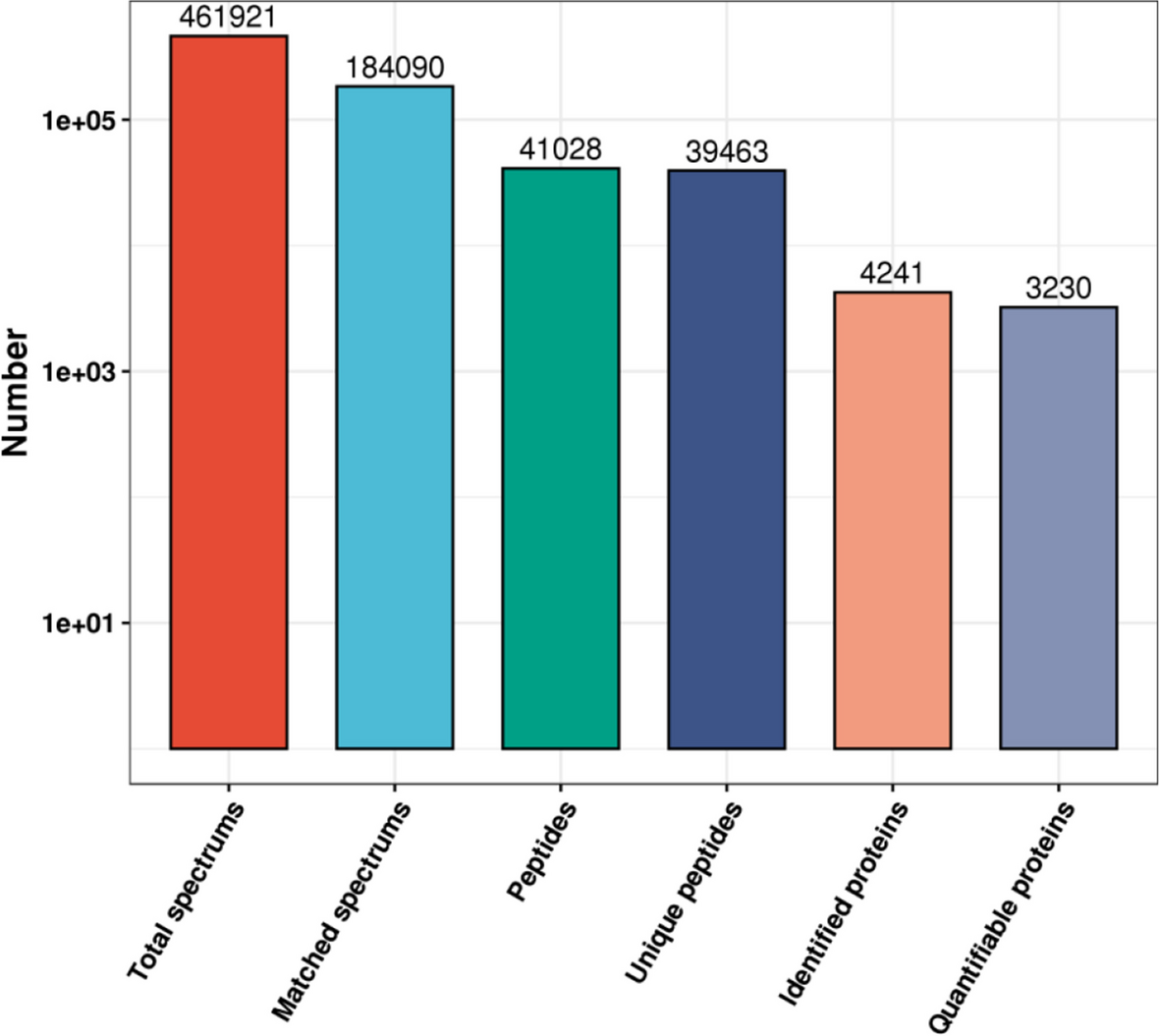
Raw files were analyzed by the Proteome Discoverer software version. 2.4 (Thermo Scientific) using the SEQUEST, Percolator, and Minora algorithms. LC–MS spectrum was matched against the UniProtKB reviewed database (11/05/2021). For protein identification and quantification, the setting parameters were as follows: a maximum of two trypsin missed cleavages were allowed with a precursor mass tolerance of 20 ppm and fragment mass tolerance of 0.01 Da. Carbamidomethylation + 57.021 Da (Cysteine) was selected as static modifications and oxidation + 15.995 Da (Methionine) was selected as dynamic modifications. The Fase discovery rate (FDR) of peptide and protein identification were both set to 0.05. The normalization of relative protein abundances ratio was performed by total peptide amount for each LC-runs (across all runs; n = 18) by normalization algorithm of Proteome discoverer software. To assembly differential expressed protein list, multiple consensus workflows were used within the Proteome Discoverer software to assemble the PSMs into peptide groups, protein database matches, and finally, non-redundant proteins groups using the principle of strict parsimony as defined by the vendor software defaults. The proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD033149 and 10.6019/PXD033149 [21]. Quality control (QC) assessment including variation in TIC and peptide–spectrum match efficiency was evaluated [22]. The number of PSM was estimated by FDR values. The data passed the QC were subjected to downstream analysis.
Statistical analysis
Data are presented as the mean ± SEM. Statistical significance was analyzed by one-way ANOVA using the GraphPad Prism software (version 7.0, CA, USA). All experiments were carried out with three independent replicates (n = 3). One-way analysis of variance (one-way ANOVA) was performed using PD for proteomics software. The significance in differences was determined by Duncan’s multiple range test (p < 0.05).








留言 (0)