
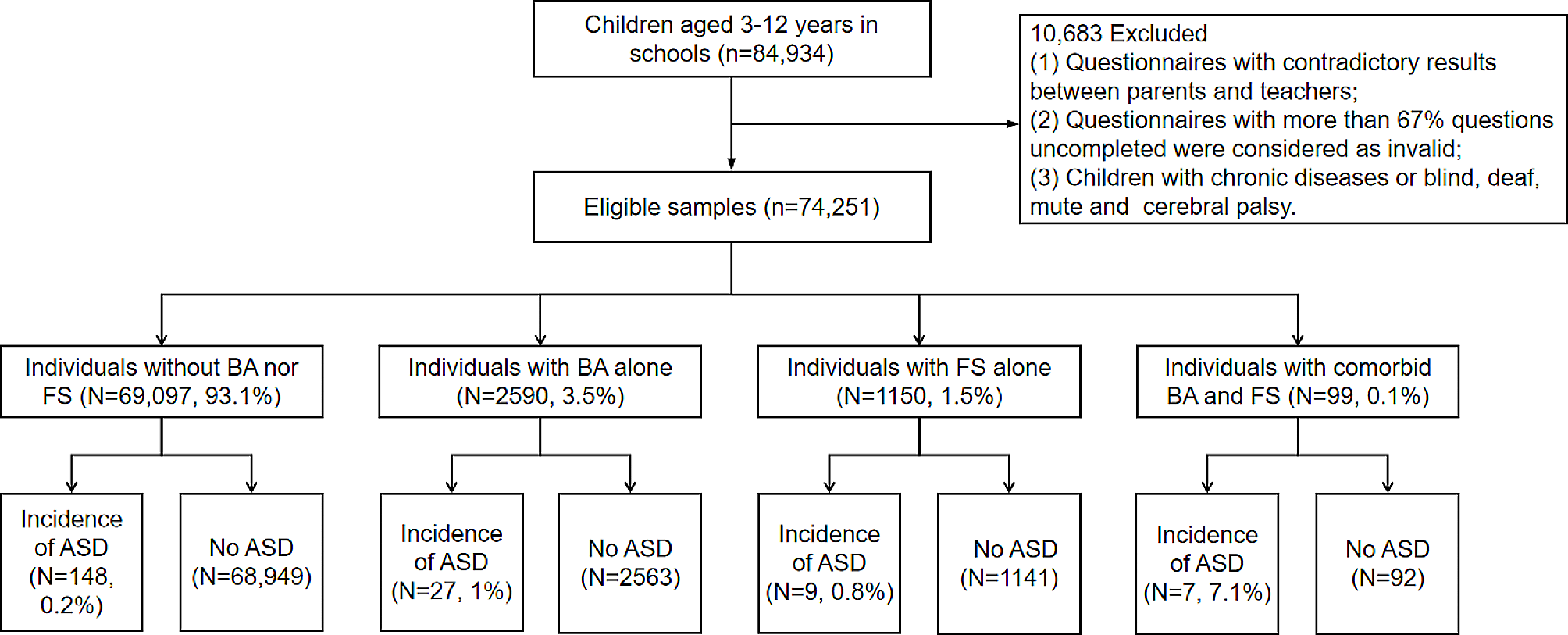
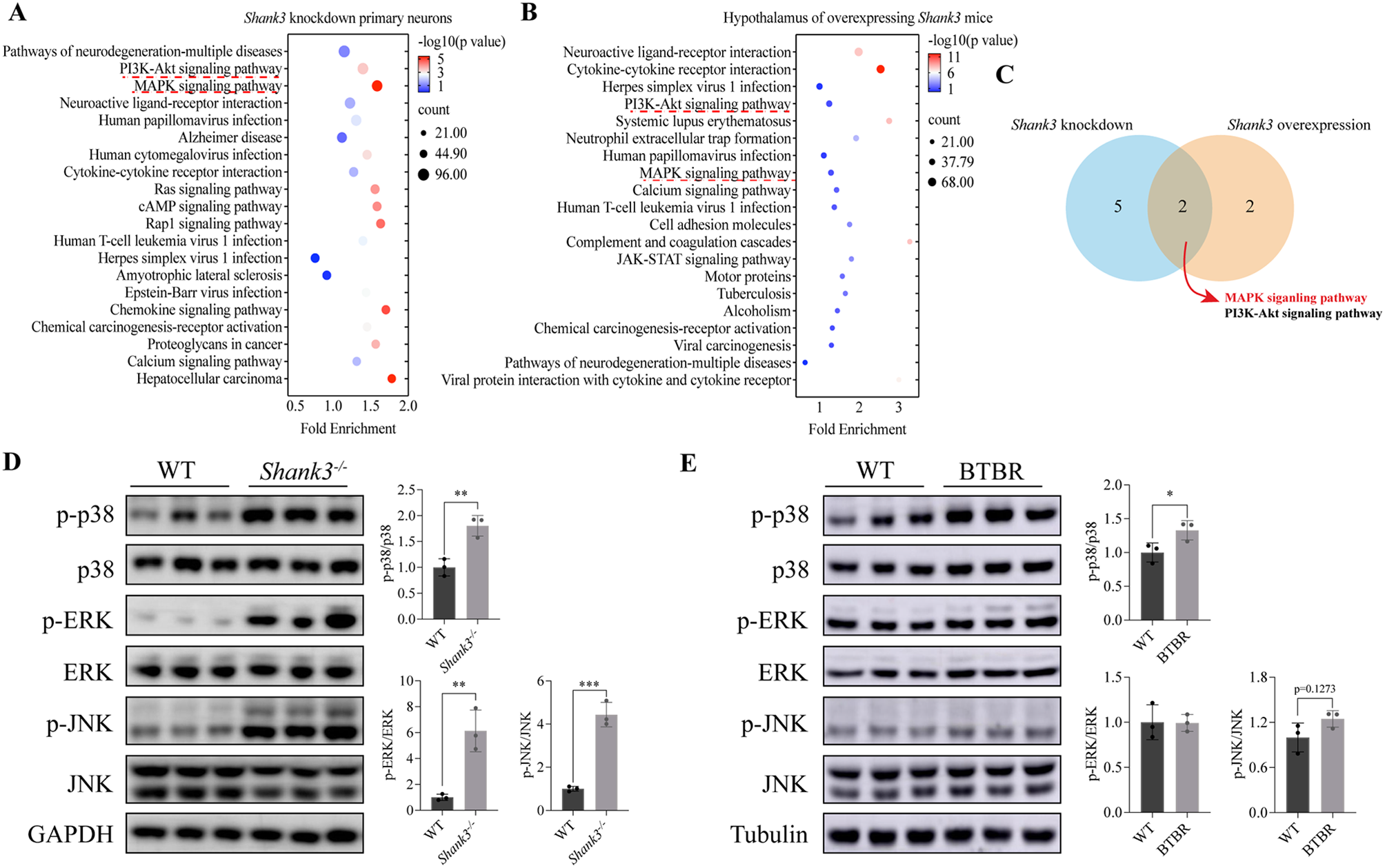
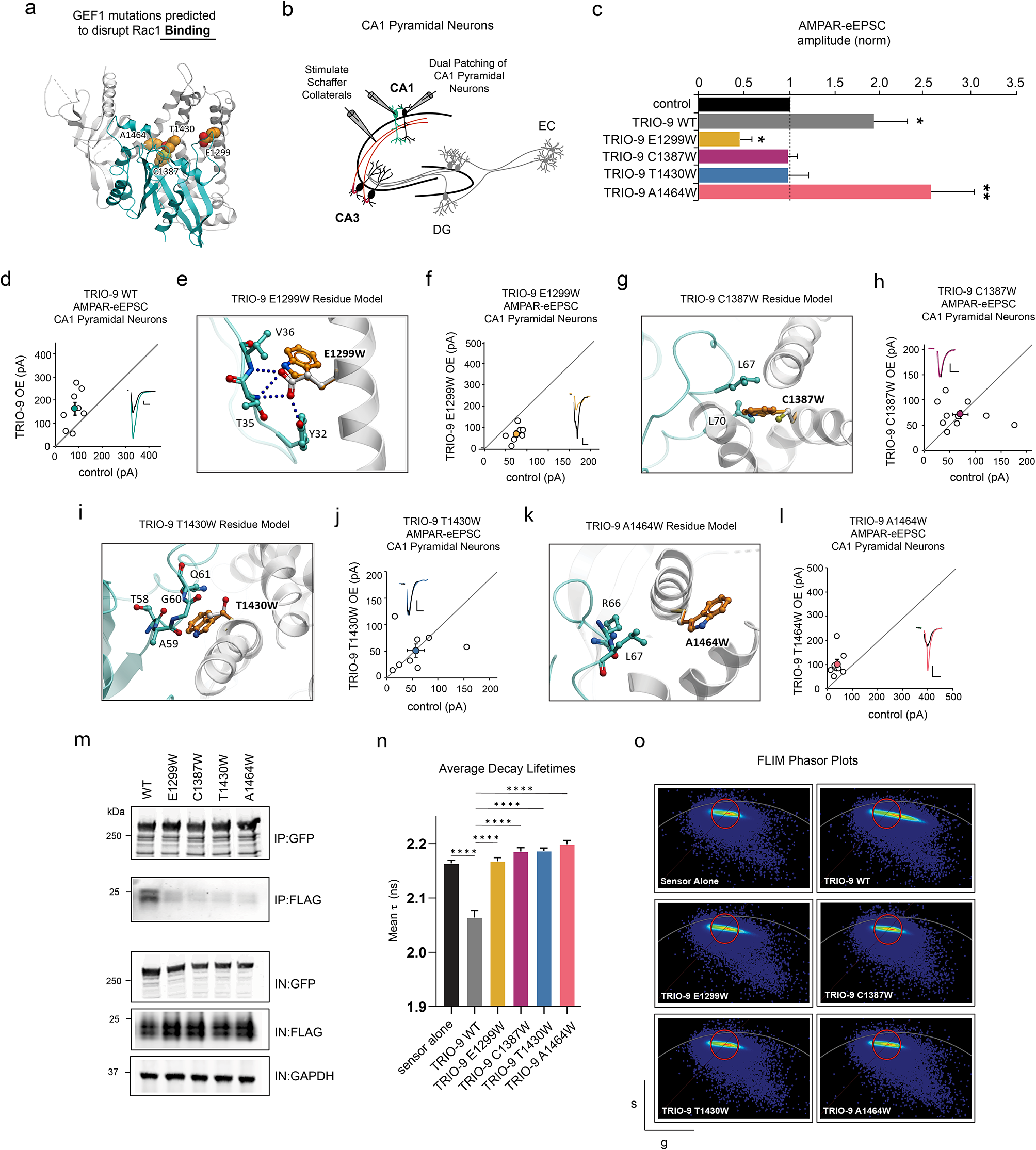
記住我



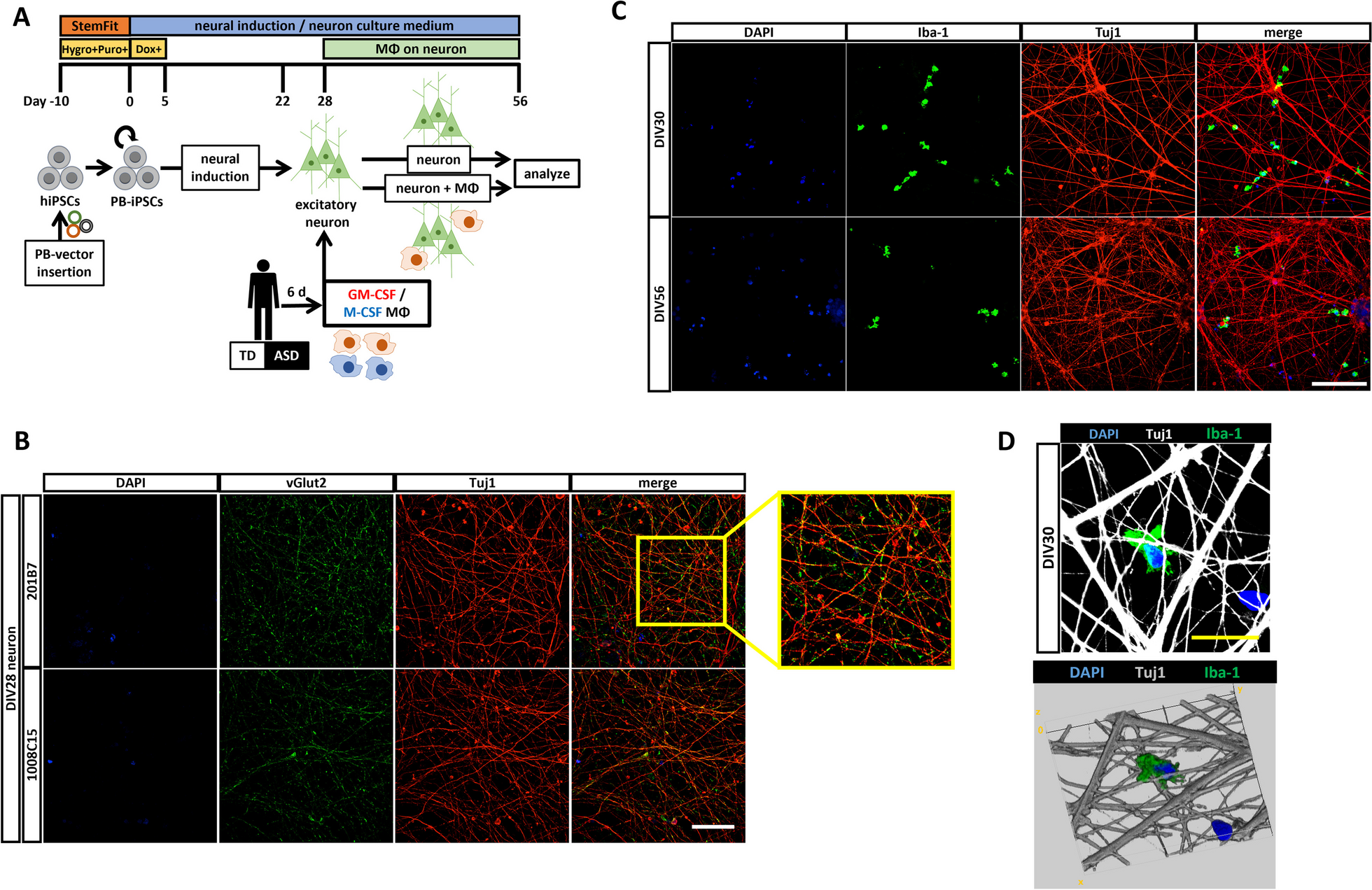



Constitutive heterozygous Myt1l+/- mutant mice were created by CRISPR/Cas9 targeting Myt1l. To generate the mutant mice, zygotes were isolated and electroporated with Cas9 proteins complexed with a guide sequence directed at exon 6, the first coding exon of the Myt1l gene. Transfected blastocysts were transferred into oviducts of day 0.5 pseudo-pregnant recipient ICR mice. Resulting pups were genotyped by Sanger sequencing of the PCR-amplified Myt1l region around the predicted cut site and several Myt1l indel mutations detected. We observed up to three different mutations in one animal suggesting continued Cas9 activity at least until the 4-cell stage. We focused on a Myt1l 7 base pair (bp) deletion allele because it is predicted to cause a frameshift and thus null allele. Mice containing this deletion 7 allele were bred with wild-type mice to achieve segregation of the various alleles and mice containing only wild-type and deletion 7 were obtained, which were used to start a colony. These Myt1l+/- mice were maintained as heterozygotes on a C57BL6/N background strain by breeding to C57BL6/N (Charles River; Strain Code: 027) mice every 4–5 months. Experimental mice were bred and housed in facilities at the Lorry I. Lokey Stem Cell Research Building.
GenotypingPCR to detect Myt1l frameshift deletion was performed with 50–200 ng of DNA extracted with QuickExtract DNA Extraction Solution (Lucigen, Middleton, WI, USA) from tail biopsies, using Taq polymerase in the Bio-Rad T100 Thermal Cycler. PCR for Myt1l deletion was performed using a forward primer for the wild-type allele F1, a separate forward primer that incorporates the deletion allele F2, and a common reverse primer R1 (see also Fig. 1A). The primers were as follows: Myt1l wild-type forward CTG AGG AGA AGC GCC ATC GCA; Myt1l deletion forward CTG AGG AGA AGC GCC ACG GT; and reverse CAC TGG TAC TCT TCT TCC ACG GAA AAT TAC C.
Fig. 1
Generation of Myt1l mutant mice by CRISPR/Cas9 gene editing. A Schematic of the gene editing strategy. The guide RNA sequence (blue text) targets seven base pairs downstream of the translation start site in exon VI of the mouse Myt1l locus and generates a frameshift deletion. The two forward and one reverse primers (red arrows) used for genotyping are noted. The forward primers, F1 and F2, incorporate the presence and absence of the seven base pairs, respectively. B PCR genotyping of Myt1l mutant mice. As expected, the primer pair F1 and R1 (left lanes) shows a PCR amplification of only wild-type alleles, whereas the primer pair F2 and R1 (right lanes) amplifies only the deletion allele, allowing unequivocal genotyping of wild-type Myt1l+/+, heterozygous Myt1l+/-, and homozygous Myt1l−/− mice. (C) Immunoblotting of Myt1l in WT, HET, and KO E18.5 brains. Whole brain lysates were subjected to Western blotting using two different Myt1l antibodies with similar results. MAP2 was used as a loading control. Note with both antibodies an extra band(*) appeared in mice carrying the mutant allele corresponding to a Myt1l-related protein of ~ 158 kDa
Breeding scheme for embryonic neurobiological characterizationNeurobiological effects of Myt1l deletion were assessed by comparing male and female constitutive homozygous Myt1l−/− mutant mice to wild-type Myt1l+/+ and heterozygous Myt1l+/- littermate controls. To generate the experimental cohort, pairs of male and female heterozygous Myt1l+/- mice were set up in the evening and females were checked for plugs the following morning.
Dissection and immunoblottingWhole mouse brains were dissected from E18.5 mice. Tissue was homogenized in ice-cold cell lysis buffer consisting of 0.5% Tween-20, 50 mM Tris pH 7.5, 2 mM EDTA, and 1 mM DTT with protease inhibitors. The lysate was incubated on ice for 15 min. Nuclei were pelleted by centrifugation at 3200 rpm for 1 min at 4 °C, and the pellet was resuspended in NP-40 lysis buffer consisting of 0.5% NP-40, 50 mM Tris pH 8.0, 150 mM NaCl, 2 mM EDTA, and 1 mM DTT with protease inhibitors. Samples were spun at 14,000 rpm for 10 min at 4 °C, and the supernatant was pipetted into a new tube. Western blot was run as described [39].
qRT-PCRRNA was isolated using Trizol (Invitrogen) and RNA Clean & Concentrator (Zymo) and reverse-transcribed with Superscript III (Invitrogen). mRNA levels were quantified by real-time PCR assay using SYBRGreen (Thermo Fisher Scientific) and the Applied Biosystems QuantStudio 7 Pro Real-Time PCR system. Expression values were expressed as percent of MAP2 using the formula: 2-CT (target mRNA)/2-CT (housekeeping mRNA) × 100. Primers used: MAP2-F: CGGTCTCCAGGGATGAAGTG; MAP2-R: ACTTGCTGCTGTGGTTTTCC; Myt1l-F: ATGTTCCCACAACCACACCA; Myt1l-R: TACCGCTTGGCATCGTCATA.
AntibodiesThe following antibodies were used for western blot: rabbit anti-Myt1l (1:500; Millipore ABE2915) and mouse anti-β-Actin (1:10,000; Sigma A5441). Rabbit anti-Tbr2 (1:500; ab23345), rat anti-Ctip2 (1:500; ab18465), and rabbit anti-Sox2 (1:250; Millipore AB5603) were used for immunofluorescence.
Histology in embryonic tissueThe morning the plug was observed was designated embryonic day 0.5. Embryos from deeply anesthetized pregnant dams were collected at E15.5 or E18.5. Uterine horns were placed in ice-cold phosphate-buffered saline (PBS). The heads of the embryos were drop-fixed in 4% paraformaldehyde (PFA) for 3 h, cryoprotected in 30% sucrose overnight, fresh frozen in powdered dry ice, embedded in O.C.T. Compound (VWR), and stored at − 80 °C until use. 40 µm free-floating coronal sections were cut using Leica CM3050 S into 1X phosphate buffer (P.B.) containing 0.02% sodium azide. Thionin staining was performed as described [40]. For immunofluorescence, sections were incubated in 10 mM sodium citrate, pH 6.0 for 10 min at 85 °C for antigen retrieval. They were washed three times with 1X PBS and then blocked for 1 h with 1X PBS containing 0.3% Triton X-100 (Tx-100) and 5% normal goat serum (NGS). Sections were incubated with primary antibody overnight at 4 °C. On the following day, after three washes in 1X PBS, sections were incubated in the appropriate Alexa Fluor secondary antibodies (Molecular Probes) diluted 1:1,000 in blocking solution for 2 h at room temperature. The sections were counterstained with DAPI in PBS for 10 min, washed three times in 1X PBS, mounted on slides, and cover-slipped (Antifade Gold, Life Technologies).
Quantitative image analysisBrain anatomy and immunofluorescent labeling were assessed using three mouse triplets (Myt1l+/+, Myt1l+/-, and Myt1−/−) from three different litters. Nissl staining embryos at age E18.5 were analyzed in slices corresponding to sections 125 (rostral) and 155 (caudal) of the Allen Atlas of the Developing Mouse Brain. Images were taken on an Olympus Slide Scanner VS200 using the 20 × objective. In the rostral section, we measured the diameter of the cortical plates (CP) at motor cortex and barrel cortex positions, the diameter of the corpus callosum, as well as the area of striatum and septum. In the caudal section, we measured the diameter of the pallium at motor cortex and barrel cortex positions, the diameter of the hippocampus from the alveus-CA1-dentate-gyrus, as well as the area of thalamus and hypothalamus.
Immunofluorescence was imaged using a Nikon A1R laser scanning confocal and 40 × oil immersion objectives. Sox2 expression was assessed by measuring the area of the Sox2-positive signal in a 100 × 100 µm square region of the ventricular zone (VZ) (age E15.5, primary motor cortex). The borders of the VZ and CP were determined using the Sox2 and DAPI signals. Trb2 expression and Ctip2 expression were used to measure the diameters of the proximal VZ (Trb2-negative), the distal VZ (Tbr2-positive and Tbr2-negative cells), the multipolar cell accumulation zone (MAZ, part of subventricular zone, all cells Tbr2-positive), the distal subventricular zone (some isolated Tbr2-positive cells), the intermediate zone (no Tbr2 signal, no/weak Ctip2 signal), the CP (band of strong Ctip2 signal), and the marginal zone. Ctip2 positive cells were counted in a 50 × 100 µm rectangle in the CP.
General overview on behavioral phenotypingBehavioral effects of Myt1l haploinsufficiency were tested by comparing male and female constitutive heterozygous Myt1l+/- mutant mice to wild-type Myt1l+/+ littermate controls. C57BL6/N dams were bred to Myt1l+/- sires to generate the experimental cohort. The day of birth was defined as postnatal day (PND) 0. In order to avoid litter effects, only litters with both genotypes were included in the experiments. After weaning on PND 21, same-sex littermates of mixed genotypes were socially housed in groups of 2–5 mice in individually ventilated cages with corn cob bedding (Universal Euro II Type Long; 522.6 cm2 floor space, 5653.5 cm3 living space, 12.7 cm height; Innocage; Innovive, San Diego, CA, USA). Mice were maintained on a 12-h light/dark cycle and were provided food (irradiated laboratory animal diet, 18% protein; Teklad; Madison, WI, USA) and water ad libitum. Mice were identified by paw tattoo, using non-toxic animal tattoo ink (Ketchum Green Animal Tattoo Ink Paste, Ketchum Manufacturing Inc., Brockville, Canada). The ink was gently inserted subcutaneously through a 30 gauge hypodermic needle tip into the center of the paw at PND 10. Mouse tail snips for genotyping were collected by dissecting ~ 0.3 cm of tail the day of tattooing.
A comprehensive, longitudinal behavioral phenotyping approach was applied, including behavioral assays relevant to all human ASD core symptoms [41,42,43]. For behavioral phenotyping, mice of N = 7 litters (8 ± 0.38 pups/litter; mean ± SEM) were tested for sensory and motor abilities as well as isolation-induced ultrasonic vocalizations in the homing test at PND 10. After weaning at PND 21, they were tested in a battery of behavioral assays in the following order: Activity box, open field, elevated plus maze, Y-maze, social approach in the three-chamber assay, direct reciprocal social interaction, accelerated rotarod, nest building, and repetitive behavior were performed at the age of 2–4 months. Spatial learning and reversal learning, acoustic startle and pre-pulse inhibition of acoustic startle, and fear conditioning were performed at the age of 6–12 months. Mice of the behavioral cohort were also used for determining the body weight gain trajectory. Body weight was measured after every behavioral assay and every 3 weeks for up to 1 year starting from PND 42. Body temperature was measured at 14 months. IPTT-300 transponders (Bio Medic Data Systems, Seaford, Delaware, USA) were injected subcutaneously and longitudinally above the shoulder. Mice were monitored for infection and allowed to recover over 3 weeks, after which the body temperature was read with the IPTT 5515 handheld reader by holding it over a freely moving mouse (DAS-7007S; Bio Medic Data Systems, Seaford, Delaware, USA). The behavioral cohort consisted of N = 30 Myt1l+/- mice (females: N = 12; males: N = 18) and N = 26 Myt1l+/+ littermate controls (females: N = 9; males: N = 17).
All behavioral experiments were carried out between 7 am and 7 pm during the light phase of the 12-h light/dark cycle. All behavioral assays were conducted and analyzed blind to genotype.
Homing test and isolation-induced ultrasonic vocalizationsFor assessing sensory and motor abilities as well as isolation-induced ultrasonic vocalizations, pups were isolated from their mother and littermates for 10 min at room temperature (21–23 °C), using a modified protocol [44]. Individual pups were transferred to a cage with corn cob bedding (Universal Euro II Type Long; Innocage; Innovive). A new cage with clean bedding was used for each test to avoid olfactory cues. Soiled corn cob bedding from the home cage was evenly spread on one side (1/3 of the cage, nest area), while the rest of the cage was covered with clean bedding (2/3 of the cage, clean area). The soiled zone was counterbalanced across individual mouse pups. The pup was placed in the middle of the cage and video-recorded for 10 min. The floor of the arena was virtually subdivided into three zones, i.e., soiled nest zone, clean center zone, and clean no nest zone, to allow behavioral scoring using The Observer XT 11 software (Noldus, Wageningen, The Netherlands). Behavioral scoring included locomotor activity by counting line crossings. Homing performance was scored as latency to reach and the time spent in the soiled nest zone. Emission of isolation-induced ultrasonic vocalizations was monitored by an UltraSoundGate Condenser Microphone CM16 sensitive to frequencies of 15–180 kHz (flat frequency response between 25 and 140 kHz; ± 6 dB; Avisoft Bioacoustics, Berlin, Germany). The microphone was placed in the roof of the cage lid, ~ 12 cm above the floor. The microphone was connected via an UltraSoundGate 416 USG audio device (Avisoft Bioacoustics) to a computer, where acoustic data were recorded with a sampling rate of 250,000 Hz (16 bit) by Avisoft RECORDER (version 2.97). For acoustic analysis, recordings were transferred to Avisoft SASLab Pro (version 5.2.12) and a fast Fourier transform was conducted (512 FFT length, 100% frame, Hamming window, and 75% time window overlap), resulting in spectrograms with 488 Hz of frequency and 0.512 ms of time resolution. Call detection was provided by an automatic threshold-based algorithm (amplitude threshold -65 dB; hold time 10 ms; high-pass filter 30 kHz). Accuracy of call detection was verified by an experienced user. When necessary, missed calls were marked manually to be included in the automatic parameter analysis. Total numbers of isolation-induced ultrasonic vocalizations were calculated for the entire session and in 60 s time bins to visualize the time course of the ultrasonic vocalization response. Additional parameters included latency to start calling, call duration, peak frequency, peak amplitude, and frequency modulation. Finally, the temporal organization of isolation-induced ultrasonic vocalizations emission was assessed through sequential analyses and call subtypes were determined by means of density plots. After the 10-min isolation period, body temperature and weight were determined. For body temperature determination, a Testo 110 thermometer with surface sensor (Testo AG, Lenzkirch, Germany) was used. Body weight was measured using a palmscale (PS7-200; precision: 0.01 g; MyWeigh Europe, Hückelhoven, Germany).
Activity boxLocomotor activity, exploratory behavior, and anxiety-related behavior were assessed under direct white light (~ 20 lx) in an activity box (ENV-510, 27.31 × 27.31 × 20.32 cm; Med Associates, Fairfax, VT, USA), using a modified protocol [45]. The activity box was housed within a sound-attenuating chamber, equipped with a ventilation fan and illuminated by a single overhead light. Mice were allowed to freely explore the activity box on two consecutive days for 30 min each day. The position of the mouse within the arena was tracked in three dimensions by arrays of infrared light beams connected to a computer running Activity Monitor software (Med Associates, Fairfax, VT, USA). This software was used to calculate distance traveled and the number of rearings during 1-min time bins, which were summed together to calculate total values throughout the entire 30-min test session. The activity box was thoroughly cleaned between each mouse using 70% ethanol to avoid olfactory cues.
Open fieldLocomotor activity, exploratory behavior, and anxiety-related behavior were also measured under more anxiogenic conditions under direct bright white light (~ 200 lx) in a white open field (34 × 34 × 40 cm), using a modified protocol [46]. At the beginning of the test, individual mice were placed into one corner of the open field. Mice were allowed to freely explore the open field for 10 min. Distance traveled and time spent in the center were recorded and analyzed using Viewer III tracking software (Biobserve, Bonn, Germany). The center area was defined as the 28 × 28 cm central section of the open field. Fecal boli were counted at the end of test session. The open field was thoroughly cleaned between each mouse using 70% ethanol to avoid olfactory cues.
Elevated plus mazeAnxiety-related behavior in the elevated plus maze was measured under indirect white light (~ 50 lx), using a modified protocol [46]. The gray maze was elevated 50 cm above the floor and consisted of four arms, i.e., two open arms and two closed arms with 15-cm-high walls, each arm measuring 35 cm long and 5 cm wide. Individual mice were initially placed in the center of the maze, facing an open arm. Mice were then allowed to freely explore the maze for 5 min. The amount of time spent in each arm, number of entries into each arm, total distance traveled, and average velocity were recorded and analyzed using Viewer III tracking software (Biobserve, Bonn, Germany). The elevated plus maze was thoroughly cleaned between each mouse using 70% ethanol to avoid olfactory cues.
Y-mazeSpatial working memory in the Y-maze was measured under indirect red light conditions, using a modified protocol [46]. A gray plastic Y-maze was used to evaluate spontaneous alternations reflecting spatial working memory. The maze consisted of three arms that were spaced 120° angle from each other (dimensions of each arm 40 × 10 × 17 cm). Mice were individually placed in the distal end of one arm and allowed to freely explore the entire maze for 10 min. A completed arm entry was defined as the entering of the mouse with all four limbs. The sequence of arm entries was recorded and analyzed using the Viewer III tracking system (Biobserve, Bonn, Germany). Visiting all three different arms consecutively was termed a ‘correct’ triad, and visiting one arm twice in three consecutive entries was termed a ‘wrong’ triad. Correct alternation percentage was calculated using the following formula: %Alternation = (Number of Alternations/[Total number of arm entries − 2]) × 100. The Y-maze was thoroughly cleaned between each mouse using 70% ethanol to avoid olfactory cues.
Social approach assaySocial motivation was evaluated in a three-chamber box (60 × 30 × 30 cm3) made of transparent polycarbonate under indirect red light conditions, using a modified protocol [47]. Retractable doorways built into the two dividing walls controlled access to the side chambers. The test session began with a 10-min habituation session during which lack of an innate side preference was confirmed. The subject mouse was then briefly confined to the center chamber, while a clean novel object, an empty metal enclosure, was placed in one of the side chambers. A novel stimulus mouse previously habituated to the enclosure was placed in an identical metal enclosure located in the other side chamber. The side containing the novel object and the novel stimulus mouse alternated between the left and right chambers across subject mice. After both stimuli were positioned, the two side doors were simultaneously lifted and the subject mouse was allowed access to all three chambers for 10 min. C57BL6/N mice served as stimulus mice. Stimulus mice were age-matched and of the same sex as the subject mouse. The amount of time spent exploring the metal enclosures, the amount of time spent in each chamber, number of entries into each chamber, and total distance traveled were recorded and analyzed using Viewer III tracking software (Biobserve, Bonn, Germany). The three-chamber box and the metal enclosures were thoroughly cleaned between each mouse using 70% ethanol to avoid olfactory cues.
Interaction-induced ultrasonic vocalizationsEmission of interaction-induced ultrasonic vocalizations was assessed during direct reciprocal social interaction in a test chamber (50 × 25 × 30 cm3) made of transparent polycarbonate under indirect red light conditions, using a modified protocol [48]. A transparent polycarbonate lid containing 16 holes (diameter 1.3 cm) was placed on the top of the social interaction chamber to reduce background noise. Clean corn cob bedding was evenly spread on the floor. Male–female and female–female pairs of mice were allowed to socially interact for 5 min after the subject mouse was habituated to the test environment for 1 min. Age-matched C57BL6/N mice served as stimulus mice. Stimulus mice were tail-marked. Interaction-induced ultrasonic vocalizations emitted by pairs were monitored by an UltraSoundGate Condenser Microphone CM16 (Avisoft Bioacoustics) placed in the roof of the lid, ~ 30 cm above the floor. The microphone was connected via an UltraSoundGate 416 USG audio device (Avisoft Bioacoustics) to a computer, where acoustic data were recorded with a sampling rate of 250,000 Hz (16 bit) by Avisoft RECORDER (version 2.97). For acoustic analysis, recordings were transferred to Avisoft SASLab Pro (version 5.2.12) and a fast Fourier transform was conducted (512 FFT length, 100% frame, Hamming window, and 75% time window overlap), resulting in spectrograms with 488 Hz of frequency and 0.512 ms of time resolution. An experienced user counted the number of ultrasonic vocalizations in 1-min time bins. The social interaction chamber was thoroughly cleaned between each pair of mice using 70% ethanol to avoid olfactory cues.
Accelerated rotarodMotor coordination and motor learning was tested under white room light using a five-station rotarod treadmill (ENV-575 M, Rota-Rod software; Med Associates, Fairfax, VT, USA), as previously described [45]. Testing consisted of three trials per day, separated by at least 60 min each, over the course of 4 days, i.e., 12 trials in total. On the first day of testing, mice were acclimated to the apparatus by placement on the stationary rotarod for 30 s. Two versions of the rotarod task were used. First, the standard task with an accelerating rod from 4 to 40 rpm within 300 s was applied for 2 days, i.e., 6 trials in total. Then, the standard range of acceleration from 4 to 40 rpm was expanded using custom hardware purchased from the vendor, allowing us to test an acceleration of 8–80 rpm, while maintaining a constant rate of acceleration over 300 s. The version with an acceleration of 8–80 rpm was also applied for 2 days, i.e., 6 trials in total. In both versions, the latency to fall off and the latency to make one complete revolution while hanging on were measured. A trial was stopped after 300 s (maximum speed, no further acceleration). The rotarod was thoroughly cleaned between each trial using 70% ethanol to avoid olfactory cues.
Nest buildingNest building was measured under white room light, using a modified protocol [49]. After mice were habituated for 15 min to a novel cage with corn cob bedding but no other nest material (Universal Euro II Type Long; Innocage; Innovive), a 5 × 5 cm square of pressed cotton (Nestlet; Ancare, Bellmore, NY) was placed in a random cage corner, and the net increase in nest width and nest height was measured after 30, 60, and 90 min. At the end, nest quality was scored.
Repetitive behaviorRepetitive behavior was measured under indirect white light (~ 40 lx), using a modified protocol [49]. After mice were habituated for 10 min to a novel cage with corn cob bedding but no other nest material (Universal Euro II Type Long; Innocage; Innovive), self-grooming of all body regions was recorded for 10 min from the side (Brio 4 K ultra HD webcam; Logitech Europe S.A.) and analyzed by a trained observer using The Observer XT 11 software (Noldus).
Spatial learning and reversal learningSpatial learning and reversal learning were assessed in a Barnes maze under direct bright white light (~ 200 lx), using a modified protocol [50]. The white maze consisted of a brightly lit circular open platform (92 cm diameter) with 20 equally spaced holes (5 cm diameter) along the perimeter. Underneath the designated target hole, an escape box (7 cm deep, 7 cm width, and 10 cm length) was placed. Underneath the remaining holes, false escape boxes were placed, made of the same material as the escape box. Extra-maze cues were placed on the surrounding walls to serve as reference cues to learn the position of the target escape hole. At the beginning of the test, individual mice were placed in the center of the maze in a holding chamber (15 × 15 cm) for 30 s. Once the chamber was lifted, the mice were allowed to freely explore the maze for 90 s with 19 of the 20 holes closed and were assayed for their ability to spatially navigate the maze to find the target escape hole. The target escape box was positioned underneath the maze as a small, dark, recessed chamber, which the mice naturally sought out, taking advantage of their desire to escape brightly lit and exposed environments. During the initial, consecutive four-day training period, the mice learned the spatial location of the target hole, with four trials conducted per day (~ 2 h inter-trial interval). For reversal learning, the target hole was rotated by ~ 180°. The mice were again trained for four consecutive days, with four trials conducted per day (~ 2 h inter-trial interval). The maze was thoroughly cleaned between each trial using 70% ethanol to avoid olfactory cues. Data acquisition and analysis were performed using the Viewer III tracking system (Biobserve, Bonn, Germany). During spatial learning and reversal learning, the latency needed to reach the target hole was measured. During the last spatial learning day, the number of visits of the target hole and adjacent holes was quantified and used as a preference measure. Similarly, the number of visits of the initial target hole, the reversal target hole, and adjacent holes was quantified during the first reversal learning day. Affinity was determined by measuring the time spent within the initial target hole and the time spent within the reversal target hole. For hole visit and affinity, an entry was defined as the mouse entering the hole with the snout or major parts of the body. Primary errors were also calculated as entering the wrong hole before reaching the target hole.
Acoustic startle reactivity and pre-pulse inhibition of acoustic startleAcoustic startle reactivity and pre-pulse inhibition of acoustic startle were measured using the Kinder Scientific startle reflex system (Kinder Scientific, Poway, CA, USA), as previously described [46]. Data were analyzed with the Startle Monitor II software (Kinder Scientific). Mice were individually placed in a small cage atop a force plate within a sound attenuation chamber without light. Background noise was set at 65 decibel (dB). The startle response, defined as the change in amplitude of force in response to an unexpected acoustic stimulus, was measured. The peak values of the absolute force mice placed on the bottom of the cage were measured as the startle response. For the acoustic startle reactivity experiment, 50 ms noise at 75, 85, 95, 105, and 115 dB was presented. Each stimulus was repeated 10 times. For the pre-pulse inhibition experiments, 50 ms noise at 115 dB was presented, with preceding noise at 0, 68, 71, or 77 dB. The acoustic startle reactivity experiment had three phases. First, the startle response was determined by presenting 10 consecutive 115 dB pulse trials. The following trials were then presented 10 times each in pseudorandom order: 115 dB pulse with 0 dB pre-pulse, 68 dB pre-pulse, 71 dB pre-pulse, and 77 dB pre-pulse. The 115 dB pulse followed each pre-pulse at a 100-ms onset–onset interval. Then, the startle response was again determined by presenting 10 consecutive 115 dB pulse trials. The percent inhibition of the startle amplitude displayed during pulse trials was calculated for each pre-pulse/ pulse pair. For both experiments, mice were given 3 min of habituation time before the sound was delivered. Stimulus sequence and inter-stimulus intervals were both pseudo-randomized. The cage was thoroughly cleaned between each trial using 70% ethanol to avoid olfactory cues.
Fear conditioningFear conditioning was conducted using the Coulbourn fear conditioning system (Coulbourn Instruments, Holliston, MA, USA), as previously described [46]. Data were analyzed with the FreezeFrame software (Actimetrics Software, Wilmette, IL, USA). On the training day, individual mice were placed in the fear conditioning chamber (18.5 × 18.5 × 21.5 cm; H10-11 M-TC; Coulbourn Instruments) outfitted with a metal grid floor and located in the center of a sound-attenuating cubicle with white house light (~ 20 lx; Coulbourn Instruments). The conditioning chamber was cleaned using 70% ethanol to provide background odor. A ventilation fan provided background noise at ~ 55 dB. As the conditioned stimulus (CS), a 2 kHz tone was presented at 85 dB for 30 s. As the unconditioned stimulus (US), a 0.75 mA foot shock was applied for 2 s through the Coulbourn precision animal shocker (Coulbourn Instruments). The foot shock co-terminated with the tone. After a 2-min habituation period, three CS–US pairings separated by 1-min inter-stimulus intervals (ISI) were delivered. Mice remained in the conditioning chamber for another 60 s before being returned to their home cages. In the context test, 24 h after training, mice were placed back into the original conditioning chamber for 5 min to assess contextual recall. The conditioning chamber was thoroughly cleaned between each mouse using 70% ethanol to avoid olfactory cues. During the altered context test, 48 h after training, the conditioning chamber was modified by changing its metal grid floor to a plastic sheet, white metal side walls to plastic walls decorated with stripes of various colors, and the background odor of 70% ethanol to 1% vanilla. Mice were placed in the altered chamber for 5 min. After this 5-min period, the CS was delivered for 1 min to assess cued recall. Freezing behavior was defined as the absence of motion lasting 1 s or longer was recorded and analyzed automatically in 30-s time bins using the FreezeFrame software.
Statistical analysisStatistical analysis was performed using SPSS (IBM Statistics SPSS, version 26, Chicago, IL, USA) and GraphPad Prism (GraphPad Software, version 9.1.0, San Diego, CA, USA). SigmaPlot (Systat Software GmbH, version 13, Erkrath, Germany), BioRender, Adobe Creative Suite 6 Photoshop and Illustrator were used to create figures. The comparison between the observed genotype distribution and expected Mendelian distribution was assessed by the chi-square goodness-of-fit test. A significant trend among the survival curves was assessed using a logrank Mantel–Cox test. For the other statistical comparisons, primarily analyses of variance (ANOVAs) were used. Specifically, to compare developmental profiles and behavioral phenotypes, ANOVAs with the between-subject factors genotype and sex or ANOVAs for repeated measurements with the between-subject factors genotype and sex and relevant within-subject factors, such as age, time, trial, or preference, were conducted. ANOVAs were followed by post hoc testing using individual ANOVAs or paired and unpaired t tests when appropriate. A p < 0.050 was considered statistically significant.
留言 (0)