
記住我


Tau and Pyk2 sequences were subcloned into an AAV-CAG-GFP vector (RRID:Addgene_28014) and GSK3β and Fyn sequences subcloned into a pcDNA3.0 vector which served as a negative transfection control.
Hek293T cell culture and transfectionHuman embryonic kidney 293T (Hek293T, RRID:CVCL_QW54) cells were cultured in DMEM (Gibco #11965092) with 10% FBS (Gibco #16000044) and incubated at a constant 37 °C with 5% CO2. For protein over-expression, Hek293T cells were transfected with appropriate DNA constructs (0.5 μg DNA/well in a 12-well plate) using Lipofectamine 3000 reagent (Invitrogen #L3000001). Cells were harvested in 1% Triton X-100 containing 50 mM Tris, 150 mM NaCl and 1 mM EDTA with protease (Roche #11836170001) and phosphatase (Roche #4906845001) inhibitors. Lysates were spun at 14,000×g for 10 min at 4 °C and Triton X-100-soluble supernatants were boiled in Laemmli sample buffer (Bio-Rad #1610747) at 95 °C for 10 min.
AnimalsB6;C3-Tg (Prnp-MAPT*P301S) PS19Vle/J (RRID:IMSR_JAX:008169) mice purchased from Jackson Laboratories (JAX) were backcrossed over 5 generations to a C57BL/6 J background to obtain hemizygous PS190/+ animals. Pyk2−/− mice, backcrossed to a C57BL/6J background over 10 generations by Schlessinger and colleagues [39] (RRID:MGI_3584536), were generously provided by Dr. David Schlaepfer (University of California–San Diego). Hemizygous PS190/+ and Pyk2+/− mice were paired to obtain WT, Pyk2−/−, PS190/+ and Pyk2−/−; PS190/+ mice. All experiments used littermate control mice with no preference for either female or male animals. Comparisons of male and female outcomes by group were conducted post hoc. All protocols including animal husbandry, genotyping, behavioral testing and euthanasia were approved by the Yale University Institutional Animal Care and Use Committee (IACUC). Animals were housed in groups of 2–5 mice per cage with access to food and water ad libitum. 12-h light/dark cycles were maintained throughout the duration of animal housing with light periods beginning consistently at 7 am.
Acute brain slice pharmacology4.5–5.5-month-old PS190/+ mice were sacrificed via live decapitation in accordance with Yale University’s Institutional Animal Care and Use Committee standards. Brains were dissected on ice and sectioned using a Leica WT1000S Vibratome in ice-cold, oxygenated (95% O2, 5% CO2) artificial cerebrospinal fluid (aCSF) containing 119 mM NaCl, 2.5 mM KCl, 2.7 mM MgSO4, 26 mM NaHCO3, 11 mM D-glucose and 1.25 mM NaH2PO4. Three 400-μm-thick coronal sections (containing the three most rostral sections of hippocampus) were collected per brain. The two hemispheres of each section were divided medially and slices enriched for hippocampal tissue by removal of the ventral half of each section using a sharp razor blade. Hippocampal-enriched brain slices were transferred to a radial-arm incubation chamber (Scientific Systems Designs Inc. #BSK6–6) containing room temperature aCSF supplemented with 2.4 mM CaCl2 and continuously oxygenated with 95% O2 and 5% CO2. After a 30 min recovery, slices were treated with either 1 μM PF-719 (Chinglu Pharmaceutical Research) or equal volume of DMSO for 2 h at room temperature. After treatment, slices were immediately homogenized on ice in 100 μl RIPA (EMD Millipore #20–188) with protease and phosphatase inhibitors (Thermo Scientific #1861281) using a polypropylene pellet pestle and spun at 100,000×g for 30 min at 4 °C. RIPA-soluble supernatants were boiled in Laemmli sample buffer with 5% β-mercaptoethanol and 1% SDS at 95 °C for 5 min.
iPSC-derived human cortical neuronsNeural induction and terminal differentiationiPSC-derived human cortical neurons were derived from zero-footprint Gibco Episomal hiPSCs (Gibco #A18945) using a previously described and validated dual SMAD inhibition protocol [40]. hiPSCs were cultured in Essential 8 Flex Medium (Gibco #A2858501) on vitronectin (Gibco #A14700)-coated plates and regularly passaged using Gentle Dissociation Medium (Stemcell Technologies #07174). When confluent, hiPSCs were dissociated using Accutase (Stemcell Technologies #07920) and re-plated at a density of 2*105 cells/well on a vitronectin-coated 12-well plate with 2 μM thiazovivin (Stemcell Technologies #72252) to improve cell-survival. One day after plating (at ~ 75% confluence), Essential 8 Flex Medium was replaced with a neural induction medium [a 1:1 mixture of DMEM/F12 (Gibco #11330) and Neurobasal-A Medium (Gibco #10888022) containing N-2 1:100 (Gibco #17502048), B-27 1:50 (Gibco #17504044), 20 μg/ml insulin (Sigma-Aldrich #I0516), 1 mM L-glutamine (Gibco #25030081), 100 μM MEM Non-Essential Amino Acids (Gibco #11140050), 0.1 mM β-mercaptoethanol (Gibco #21985), 100 nM LDN-193189 (Cayman Chemical #19396), 10 μM SB-431542 (Cayman Chemical #13031) and 2 μM XAV-939 (Tocris #3748)] replaced daily for 12 days. On day 13, cells were dissociated using Accutase and seeded onto 24-well poly-D-lysine plates (Corning #354414) additionally coated with 5 μg/ml laminin (Gibco 23,017,015) in neural induction medium with 2 μM thiazovivin at a density of 4*104 cells/well. Neural induction medium was replaced 2 to 3 days after seeding with terminal neural differentiation medium (Neurobasal-A Medium containing N-2 1:100, B-27 1:50, 1 mM L-glutamine and 30 ng/ml BDNF (Gibco #PHC7074). Cells were maintained in terminal neural differentiation medium, ¾ of which was replenished twice-weekly for more than 120 days. To prevent detachment, terminal neural differentiation medium was supplemented with 2.5 μg/ml laminin once-weekly.
hiPSC-derived neuron pharmacology1 hr prior to treatment, ¾ of medium was replaced with fresh terminal neural differentiation medium. For treatment, cells were administered either PF-719 or DMSO vehicle diluted in terminal neural differentiation medium. For each treatment condition, volumes of DMSO vehicle and DMSO-solubilized PF-719 were kept constant to control for DMSO-induced modulation of cellular signaling events. Neurons were treated for 2 h at 37 °C and, following treatment, immediately harvested on ice in 100 μl/well RIPA (from a 24-well plate) with 1% SDS and protease/phosphatase inhibitors. Samples from adjacent wells were pooled (pooled sample volume, 200 μl from 2 wells), briefly vortexed and spun at 100,000×g for 30 min at 4 °C. SDS-soluble supernatants were boiled in Laemmli sample buffer with 5% β-mercaptoethanol and 1% SDS at 95 °C for 5 min.
Brain tissue collection and processing9.5–10.5-month-old animals were sacrificed via live decapitation and hemispheres separated medially on ice using a sharp razor blade. For immunohistology, left hemispheres were post-fixed in PBS with 4% paraformaldehyde (PFA) for 48 h at 4 °C. Post-fixed hemispheres were then transferred to PBS with 0.05% sodium azide and stored at 4 °C until sectioning. For biochemistry, hippocampi and cortices were immediately dissected from right hemispheres on ice. To obtain TBS-soluble fractions, hippocampi and cortices were homogenized in 150 μl and 300 μl, respectively, TBS with protease and phosphatase inhibitors using a polypropylene pellet pestle on ice. Homogenates were spun at 100,000×g for 30 min at 4 °C. TBS-soluble supernatants were boiled in Laemmli sample buffer with 5% β-mercaptoethanol and 1% SDS at 95 °C for 5 min, while TBS-insoluble hippocampal and cortical pellets were resolubilized on ice in 150 μl and 300 μl, respectively, RIPA with 1% SDS and protease/phosphatase inhibitors. Homogenates were spun at 100,000×g for 30 min at 4 °C and SDS-soluble supernatant boiled in Laemmli sample buffer with 5% β-mercaptoethanol and 1% SDS at 95 °C for 5 min.
ImmunoblottingSamples were separated via SDS-PAGE through 4–20% Tris-glycine gels (Bio-Rad #5671095) and transferred onto nitrocellulose membranes (Invitrogen #IB23001) using an iBlot 2 Gel Transfer Device (Invitrogen #IB21001). Loaded sample volumes were normalized to total protein concentration via BCA protein assay (Thermo Scientific #23225). Nitrocellulose membranes were blocked while rocking at room temperature for 1 h (Rockland #MB-070-010TF) and incubated with blocking buffer containing primary antibodies overnight at 4 °C. The following antibodies were employed for immunoblotting: anti-Tau (HT7) (Thermo Fisher Scientific #MN1000, 1:1000, RRID:AB_2314654), anti-pTau S202/T205 (AT8) (Thermo Fisher Scientific #MN1020, 1:1000, RRID:AB_223647), anti-pTau S396/S404 (PHF-1) (Peter Davies personal request, 1:1000, RRID:AB_2315150), anti-pTau S199/S202 (Thermo Fisher Scientific #44-768G, 1:1000, RRID:AB_2533749), anti-pTau T181 (AT270) (Thermo Fisher Scientific #MN1050, 1:1000, RRID:AB_223651), anti-pTau S262 (Thermo Fisher Scientific #44-750G, 1:1000, RRID:AB_2533743), anti-Pyk2 (Cell Signaling Technology #3480, 1:1000, RRID:AB_2174093), anti-pPyk2 Y402 (Cell Signaling Technology #3291, 1:1000, RRID:AB_2300530), anti-GSK3β (Cell Signaling Technology #9315, 1:1000, RRID:AB_490890), anti-GSK3β Y216/pGSK3α Y279 (Abcam #ab68476, 1:1000, RRID:AB_10013745), anti-GSK3β S9 (Cell Signaling Technology #9336, 1:1000, RRID:AB_331405), anti-Fyn (Cell Signaling Technology #4023, 1:1000, RRID:AB_10698604), anti-pSrc Family Y216 (Cell Signaling Technology #6943, 1:1000, RRID:AB_10013641), anti-PSD-95 (Cell Signaling Technology #36233, 1:1000, RRID:AB_2721262), anti-C1q (Abcam #ab182451, 1:1000, RRID:AB_2732849), anti-p38 MAPK (Cell Signaling Technology #9212, 1:1000, RRID:AB_330713), anti-pp38 MAPK T180/Y182 (Cell Signaling Technology #4511, 1:1000, RRID:AB_2139682), anti-LKB1 (Cell Signaling Technology #3047, 1:1000, RRID:AB_2198327), anti-pLKB1 S428 (Cell Signaling Technology #3482, 1:1000, RRID:AB_2198321), anti-MAPK1 (Cell Signaling Technology #4696, RRID:AB_390780), anti-pMAPK1 T185/Y187 (Cell Signaling Technology #9101, 1:1000, RRID:AB_331646) and anti-β-Actin (Cell Signaling Technology #3700, 1:10,000, RRID:AB_2242334). For experiments shown in Fig. 1, all primary antibodies were the same as above accept for the following: anti-GSK3β (Cell Signaling Technology #12456, 1:2000, RRID:AB_2636978), anti-GSK3β Y216/pGSK3α Y279 (Abcam #ab52188, 1:1000, RRID:AB_880261), anti-Pyk2 (Cell Signaling Technology #3292, 1:1000, RRID:AB_2174097) and anti-β-Actin (Cell Signaling Technology #8457, 1:1000, RRID:AB_10950489). After primary antibody incubation, membranes were washed (3 X 5-min in TBST) and incubated with appropriate secondary antibodies for 1 h at room temperature. The following secondary antibodies were used for immunoblotting: anti-mouse IRDye 800CW (LI-COR Biosciences #926–32,212, 1:20,000, RRID:AB_621847) and α-rabbit IRDye 680CW (LI-COR Biosciences #926–68,023, 1:20,000, RRID:AB_10706167). After final washes (5 X 5-min in TBST), immunoblots were scanned with a LI-COR Odyssey infrared imaging system and protein bands quantified using LI-COR Image Studio Lite software, version 5.2.5 (RRID:SCR_013715).
Fig. 1
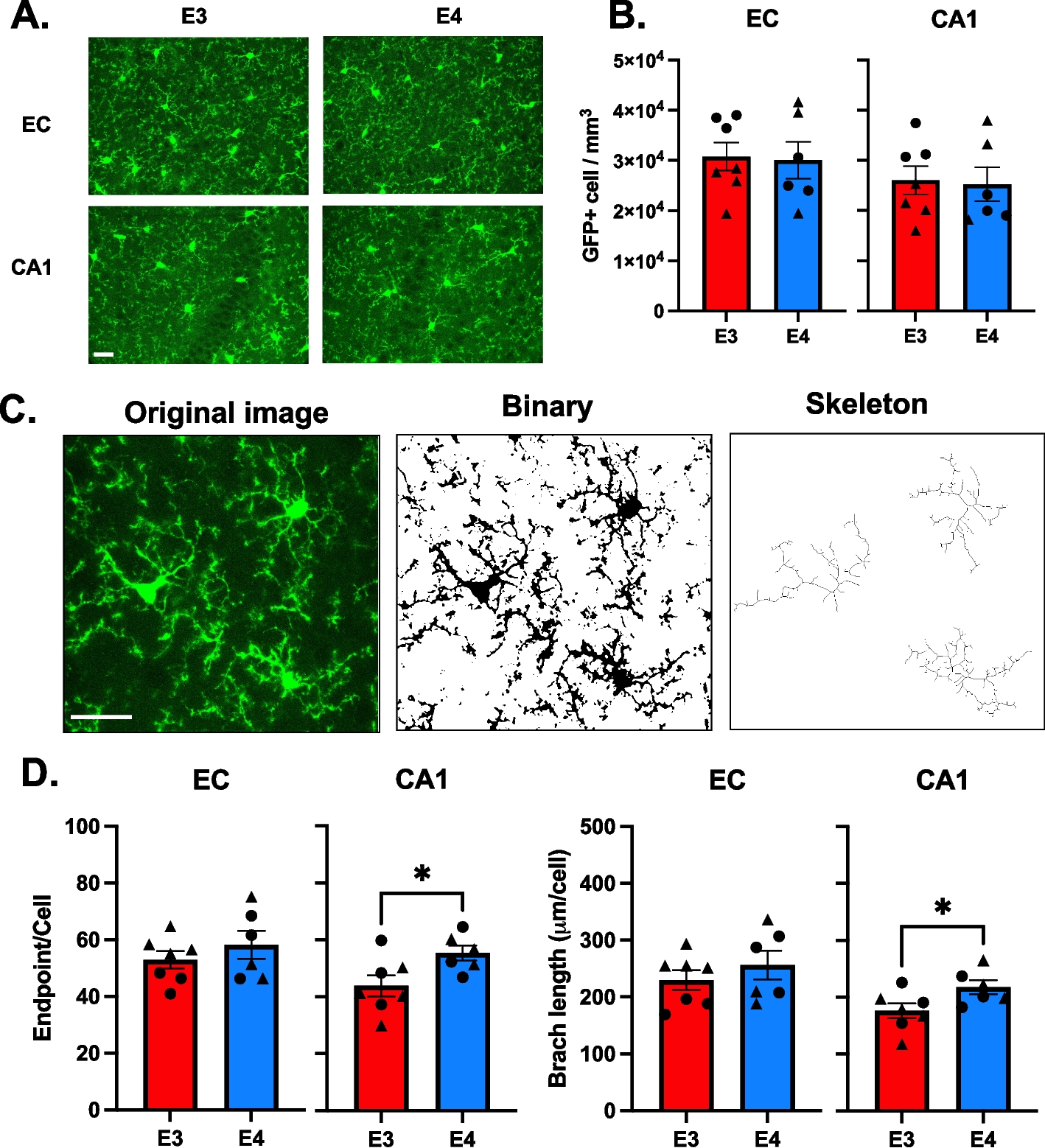
Pyk2 phosphorylates Tau via GSK3β in a Hek293T over-expression system. Hek293T cells were transfected with combinations of the proteins indicated, and lysates separated via SDS-PAGE. Separated lysates were immunoblotted with the antibodies listed. A, Representative immunoblot images of transfected Hek293T cells. B and C, Quantification of A. Over-expression of Pyk2 led to a significant increase in the activity of over-expressed GSK3β (pGSK3β Y216 normalized to total GSK3β). This increase was further augmented by the co-transfection of Fyn with GSK3β and Pyk2 (B). The phosphorylation of over-expressed Tau at S202/T205 (AT8) normalized to total Tau (HT7) was significantly increased when co-transfected with GSK3β and Pyk2, but not when co-transfected with either kinase alone. No further increase in normalized AT8 signal was observed when Tau, Pyk2 and GSK3β were co-transfected with Fyn. Data are graphed as mean ± SEM, one-way ANOVA with Tukey’s multiple comparisons test, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, n = 3
ImmunohistologyImmunofluorescenceBrains post-fixed in 4% PFA for 48 h were vibratome sectioned into 40 μm, free-floating sections, washed in PBS with 0.05% sodium azide and blocked with PBS containing 0.1% Triton X-100 (American Bio #AB02025) and 1% BSA for 1 h at room temperature. Spinal cord lumbar enlargements were post-fixed and, following embedment in gelatin, sectioned and blocked as described above. Sections were incubated in primary antibody diluted in PBS with 0.1% Triton X-100 and 1% BSA for 48 h at 4 °C. The following primary antibodies were used for immunohistology: anti-Pyk2 (Santa Cruz Biotechnology #SC-1515, 1:250, RRID:AB_632286), anti-Tau (Agilent #A0024, 1:500, RRID:AB_10013724), anti-pTau S202/T205 (AT8) (Thermo Fisher Scientific #MN1020, 1:1500, RRID:AB_223647), anti-pTau S199/S202 (Thermo Fisher Scientific #44-768G, 1:100, RRID:AB_2533749), anti-NeuN (Abcam #ab104225, 1:500, RRID:AB_10711153), anti-GFAP (Abcam #ab4674, 1:500, RRID:AB_304558), anti-Iba1 (FUJIFILM Wako Shibayagi #019–19,741, 1:250, RRID:AB_839504), anti-CD68 (Bio-Rad #MCA1957, 1:500, RRID:AB_322219), anti-PSD-95 (Thermo Fisher Scientific #51–6900, 1:250, RRID:AB_2533914) and anti-C1q (Abcam #ab182451, 1:1000, RRID:AB_2732849). Anti-PSD-95 immunolabeling required an antigen retrieval step prior to primary antibody incubation. For antigen retrieval, sections were transferred to PBS with 1% SDS and heated at 90 °C for 5 min. Following primary antibody incubation, sections were washed (3 X in PBS) and incubated in appropriate secondary antibodies diluted in PBS with 0.1% Triton X-100 and 1% BSA overnight at 4 °C. The following Alexa Fluor secondary antibodies were employed: anti-goat 488 (Thermo Fisher Scientific #A11055, 1:500, RRID:AB_2534102), anti-mouse 488 (Thermo Fisher Scientific #A-21202, 1:500, RRID:AB_141607), anti-rabbit 568 (Thermo Fisher Scientific #A10042, 1:500, RRID:AB_2534017), anti-rabbit 488 (Thermo Fisher Scientific #A-11008, 1:500, RRID:AB_143165), anti-chicken 647 (Thermo Fisher Scientific #A32933, 1:500, RRID:AB_2762845) and anti-rat 488 (Thermo Fisher Scientific #A-21208, 1:500, RRID:AB_2535794). To minimize lipofuscin autofluorescence, sections were washed after secondary antibody incubation (3 X in PBS), dipped briefly in ddH2O and incubated in ammonium acetate with 10 mM CuSO4 for 15 min at room temperature. Samples were briefly returned to ddH2O, washed in PBS and mounted onto glass slides using Vectashield mounting medium with DAPI (Vector #H-1200).
Cresyl violet stainingPost-fixed brain sections were obtained as described above and stained in filtered, pre-warmed 0.1% cresyl violet solution for 10 min. Sections were then washed in ddH2O for 3 min and de-stained in 95 and 100% ethanol for 10 and 5 min, respectively. Sections were submerged in fresh 100% ethanol for an additional 5 min, placed in xylene for 5 min, removed and placed in fresh xylene for an additional 5 min. Sections were left in xylene overnight and then mounted to glass slides with Cytoseal 60 (Thermo Fisher Scientific #8310–04).
Imaging and immunohistological analysisImage acquisitionImages of Pyk2, total Tau, phospho-Tau, NeuN, Iba1, CD68 and C1q-immunolabeled sections were captured using a Leica SP8 confocal microscope. Pyk2-immunolabeled sections were imaged using a 10X 0.4 numerical aperture air-objective lens. 4 image slices were acquired throughout the thickness of each section and z-stack compressed via maximum orthogonal projection. 12 contiguous, tiled images (3 × 4) were stitched together to image the hippocampus and cortex. Tau-immunolabeled sections were imaged with a 10X 0.4 numerical aperture air-objective lens. 20 image slices were acquired throughout the entire thickness of each section and z-stack compressed via maximum orthogonal projection. NeuN- and AT8-immunolabled spinal cord sections were imaged with a 10X 0.4 numerical aperture air-objective lens. 10 image slices were acquired throughout the thickness of each section and z-stack compressed via maximum orthogonal projection. To image the entire spinal cord lumbar enragement, 6 contiguous, tiled images (2 × 3) were stitched together. Iba1 and CD68-immunolabed sections were imaged using a 20X 0.75 numerical aperture air objective and 15 image slices, acquired throughout the thickness of each section, z-stack compressed via maximum orthogonal projection. To image the entire hippocampus, 4 contiguous, tiled images (1 × 4) were stitched together. For C1q imaging, sections were imaged with a 63X 1.4 numerical aperture oil-immersion objective and 5 image slices (1 μm apart) were z-stack compressed via maximum orthogonal projection. For PSD-95 and GFAP imaging, section images were captured using a Zeiss LSM 800 confocal microscope. PSD-95-immunolabeled sections were imaged using a 63X 1.4 numerical aperture oil-immersion objective at the z-level of greatest immunofluorescence for each section. GFAP-labeled sections were imaged with a 20X 0.8 numerical aperture air objective and 10 image slices taken throughout the thickness of the section were z-stack projected via maximum orthogonal projection. To image the entire dentate gyrus, 3 × 5 tiled images were stitched together. For all image acquisition, pinholes were set to 1 AU. Cresyl violet-stained sections were scanned using an Aperio scanner (Aperio CS2, Leica Biosystems).
Image analysisImage analysis was conducted using CellProfiler Image Analysis software, version 3.1.8 (RRID:SCR_007358). Macros to identify positive immunofluorescence signal for cell bodies or synaptic puncta were developed and applied uniformly for each immunolabeled epitope. Hippocampal cell layer thickness of cresyl violet-stained sections was determined using Aperio ImageScope, version 12.4.3.5008 (RRID:SCR_020993). All images were acquired, processed and analyzed by an experimenter blinded to animal genotype.
Behavioral assaysBehavioral assays were administered in the following order: rotarod, wire hang and Morris Water maze (MWM). Noldus CatWalk XT gait analysis was administered to a separate cohort of animals naïve to rotarod, wire hang and MWM. All animals were 9–10 months old at the time of behavioral testing. Exclusion criteria, described below, were applied independently to each assay. Animal handling and analyses (including application of exclusion criteria) were completed by an experimenter blinded to animal genotype. In order to habituate animals to experimenter handling, mice were handled for at least 2 min over 4 days prior to the first behavioral assay. For each assay, mice were habituated to the testing room for 1 h prior to behavioral testing.
RotarodAnimals were positioned on the drum of a 4-lane Rotarod device (Economex, Columbus Instruments) set to accelerate 0.1 rotations/min/sec until reaching a top speed of 4 rpm. Trials began at the start of drum rotation. Latency to fall onto a foam pad placed beneath the spinning drum was recorded across five trials with 1 min inter-trial rest periods.
Wire hangAnimals were placed in the center of a wire grid (wire spacing, 1 cm × 1 cm) which, at the beginning of each trial, was inverted over a foam pad. The latency to fall from the grid was recorded (maximum trial duration, 120 s) across two trials with a 1 min inter-trial rest period.
MWMTesting took place over 5 consecutive days, with acquisition trials taking place over the first 3 days and the probe and visible platform trials taking place on days 4 and 5, respectively. Mice were swum in an open pool ~ 1 m in diameter adorned with distinct visual cues (white poster board marked with black tape) distributed evenly along the pool perimeter. Water temperature was set to 25 °C at the beginning of each testing day. During acquisition, a clear plexiglass platform submerged 1 cm below water level was placed in the center of the target quadrant where it remained for the duration of the acquisition trials. Acquisition trials took place over 6 sessions, two sessions per day, with each session consisting of 4 trials and 1 min inter-trial rest periods. For each trial, mice were placed (facing away from the pool’s center) into the pool at one of four drop zones, the orders of which were varied across each session. The time required to locate the submerged platform was recorded for each trial using Panlab SMART video tracking software, version 2.5.21 (RRID:SCR_002852). A trial was considered successfully completed only if the animal spent 1 s on the platform. Animals unable to locate the submerged platform within the 60 s maximum trial period were gently guided to the platform and allowed to rest for 10 s. 24 h after the final acquisition trial, animals were reintroduced to the pool with the submerged platform removed. During the single probe trial, animals were placed in the zone farthest from the target quadrant and the time spent in the target quadrant recorded (maximum trial duration, 60 s). To rule out visual impairment as a potential confound, the latency required to reach a visible platform placed in the center of the pool was determined. Visible platform trials were administered until latencies stabilized (defined as a maximum range in latency of 10 s across 3 consecutive trials) over a maximum number of 15 trials, and the latencies for the final 3 trials were averaged. Animals that were unable to locate the visible platform (1 PS190/+ and 1 PS190/+;Pyk2−/− mouse), were statistical outliers (1 Pyk2−/− mouse) or whose latencies failed to stabilize over 15 trials (1 Pyk2−/− mouse) were excluded from all MWM analyses.
Noldus CatWalk XTPrior to behavioral testing, animals were habituated and trained to the CatWalk by placing their home-cage beneath a platform located at the target end of the CatWalk unit. The home-cage was accessible to the animals through an aperture in the platform. Animals were placed onto the target end platform and permitted to explore until they attempted to enter their home-cage through the platform aperture. After 3 successful attempts, a housing-unit was placed over the aperture bridging the end of the CatWalk and the home-cage entrance. Testing began after 3 successful attempts to enter the housing unit from the target end of the CatWalk. Animals were placed at the far end of the CatWalk unit and gait parameters recorded during each successful transverse of the ~ 1 m long platform track in either direction. Trials were repeated until animals achieved 3 successful runs (maximum run duration, 10 s; maximum run variation, 75%). Animals unable to ambulate across the platform track were excluded from analysis.
Label-free quantitative proteomicsSample preparationP2’ crude synaptosomal pellets from mouse brain (4 biological replicates / genotype) were homogenized by sonication in a buffer containing urea (8 M), ammonium bicarbonate (0.4 M), and protease (Pierce #87786 at 1% of lysis buffer) and phosphatase inhibitor cocktails (Pierce #78420 at 2.5% of lysis buffer). Samples were then centrifuged to pellet cellular debris and supernatant collected for downstream global proteomics sample preparation with slight modification from a previously published protocol [41]. Briefly, proteins were first extracted using cold (− 20 °C) acetone. A 1:4 ratio of protein solution to cold acetone was incubated for 1 h at − 20 °C, then centrifuged at 15,000×g to pellet out the protein precipitate. Protein pellets were reconstituted in a final solution containing 2 M urea and 25 mM ammonium bicarbonate. Cysteines were reduced with dithiothreitol (DTT) at 37 °C for 20 min, cooled and then alkylated with iodoacetamide (IAM) at room temperature in the dark for 20 min. Dual enzymatic digestion was carried out first with LysC at 37 °C for 4 h and subsequently with Trypsin overnight at 37 °C. Digestion was quenched with 0.5% formic acid. Samples were desalted using MiniSpin SPE columns (The Nest Group #HMM S18V) and dried via SpeedVac (Thermo Scientific SAVANT RVT-4104). Pellets were dissolved in a solution containing 70 mM L-glutamic acid, 65% ACN and 2% TFA in water (loading/conditioning buffer for TopTip). Samples were then subjected to titanium dioxide (TiO2) phospho-peptide enrichment using TopTip (Glygen #TT2TIO). Phospho-peptide enrichment was carried out according to manufacturing specification with the exception of the initial loading/conditioning buffer indicated above. Flow Through peptide eluate (FT, non-bound) was collected and stored at − 80 °C for mass spectrometry analysis of total proteins. Enriched phospho-peptides (EN, bound) were eluted from each TopTip in three aliquots of 30 μL 28% high-purity ammonium hydroxide. The eluted fraction was dried and re-dried with 2 × 30 μL water by SpeedVac. Enriched fractions were dissolved in 10 μL of 70% formic acid and 30 μL of 50 mM sodium phosphate. Peptide concentrations were determined by NanoDrop spectrophotometer (Thermo Scientific NanoDrop 2000) to load 0.3 μg / 5 μL of each sample for analysis (3 injections / biological replicate). For WT vs Pyk2−/− analysis, LC-MS/MS was conducted using an Orbitrap Fusion LC-MS/MS mass spectrometer equipped with a Waters nanoACQUITY Ultra Performance Liquid Chromatography (UPLC) System. For the PS190/+ vs PS190/+;Pyk2−/− analysis, LC-MS/MS was conducted using a Q Exactive HF-X Quadrupole-Orbitrap MS system.
Proteomics data analysisOnline chromatographic separation was conducted as described previously [42]. Peaks were selected and the generated peak list files were used for phospho-peptide identification using SEQUEST search algorithm in Proteome Discoverer, version 2.2 (RRID:SCR_014477). Searches were conducted against the SwissProt Protein Database (Version SwissProt_2017_01, Mus musculus) (RRID:SCR_017486). Search parameters included: fragment ion mass tolerance of 0.020 Da; parent ion tolerance of 10.0 pp.; strict trypsin fragments (enzyme cleavage after the C-terminus of Lysine or Arginine, but not if followed by Proline); variable modification of phospho- Ser, Thr, and Tyr; oxidation of Met; deamidation of Asn and Gln; and carbamidomethlyation of Cys. A decoy search (based on the reverse sequence search) was performed to estimate the false discovery rate (FDR), with a setting of acceptable protein ID having an FDR of < 1%. A protein was considered to be positively identified if there were two or more significantly labeled unique peptides. Scaffold Proteome Software, version 4.8.6 (RRID:SCR_014345) was used to obtain quantitative abundance values for comparison between WT and Pyk2−/− genotypes. Abundance values for phospho-enriched samples were normalized to total (FT) values. Proteins significantly differentiated between genotypes were identified using two-tailed t-test (p < 0.05). Two-tailed t-tests were conducted and z-scores determined using Microsoft Excel software, version 16.16.27 (RRID:SCR_016137). Enrichment of Gene Ontology terms amongst lists of DEPs were assessed using ClueGO in Cytoscape ([43], RRID: SCR_005748). A protein-protein association network between significantly differentially regulated phospho-proteins was obtained using STRING ([44], RRID:SCR_005223).
Synaptosomal fractionationAnimals were sacrificed and brain tissue dissected on ice as described above. Hippocampi and cortices from a single hemisphere were homogenized in 200 μl and 400 μl, respectively, Syn-PER Reagent (Thermo Scientific #87793) with protease and phosphatase inhibitors using a polypropylene pellet pestle. Homogenates were centrifuged at 1200×g for 10 min at 4 °C. Supernatants were collected and spun again at 15,000×g for 20 min at 4 °C. The crude synaptosomal (P2’) pellets were resolubilized in RIPA with 1% SDS and boiled in Laemmli sample buffer with 5% β-mercaptoethanol and 1% SDS at 95 °C for 5 min.
Experimental design and statistical analysisOne-way ANOVA with post hoc Tukey’s multiple comparisons tests, One-way ANOVA with post hoc Dunnett’s multiple comparisons tests, Log-rank (Mantel-Cox) test and unpaired two-tailed t-tests were performed using GraphPad Prism software, versions 8 and 9 (RRID:SCR_002798). Repeated measures ANOVA with post hoc Tukey HSD multiple comparisons tests were performed using IBM SPSS Statics software, version 26 (RRID:SCR_019096). For the MWM visible platform test, statistical outliers were identified using the ROUT method (Q = 1%) in GraphPad Prism. Group means ± SEM and samples sizes (n) are reported in each figure legend. Data were considered to be statistically significant if p < 0.05. For all figures, all statistically significant group differences are labeled. For any given group comparison, the lack of any indication of significant difference implies lack of significance by the applied statistical test.
留言 (0)