
記住我
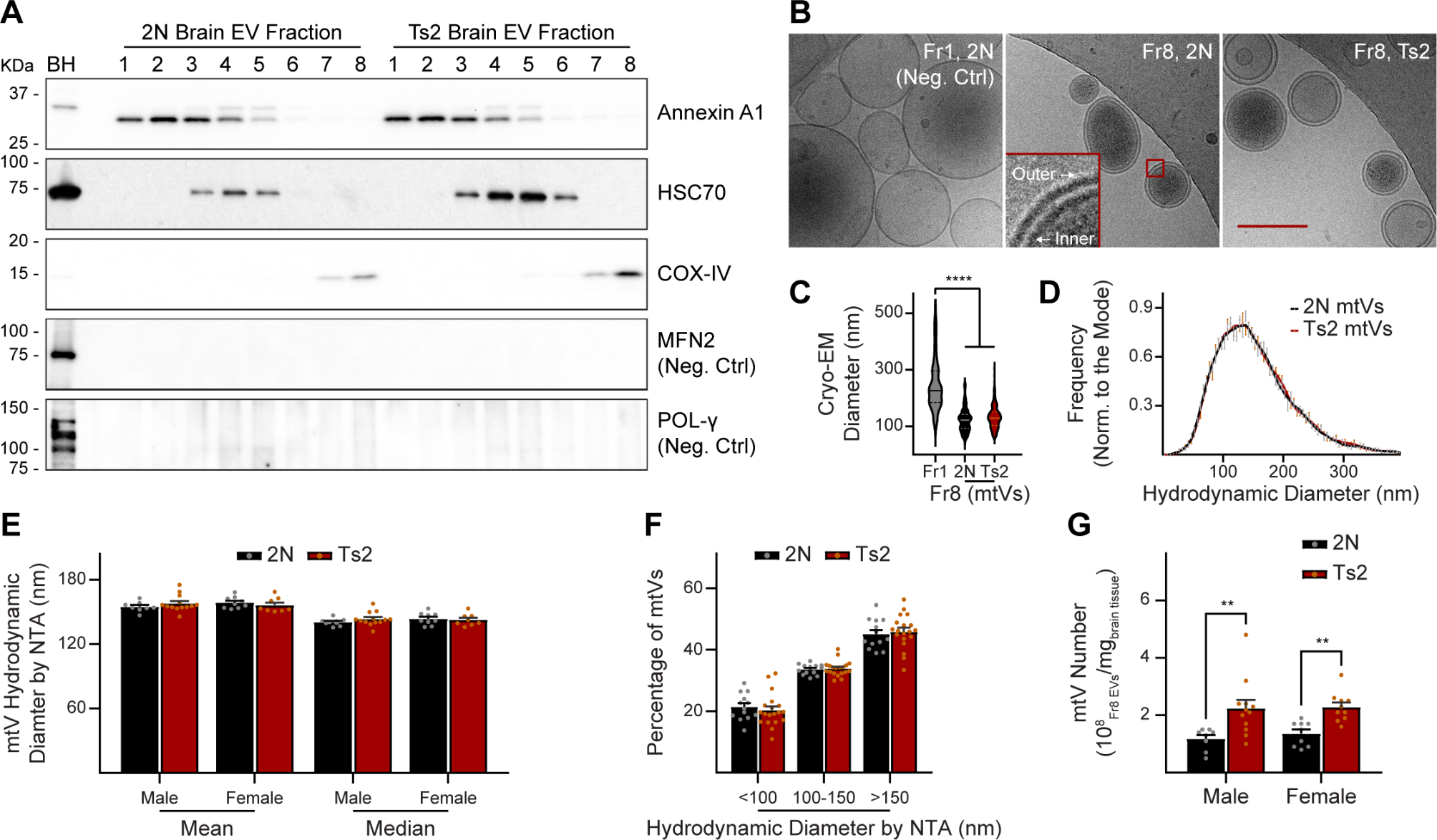

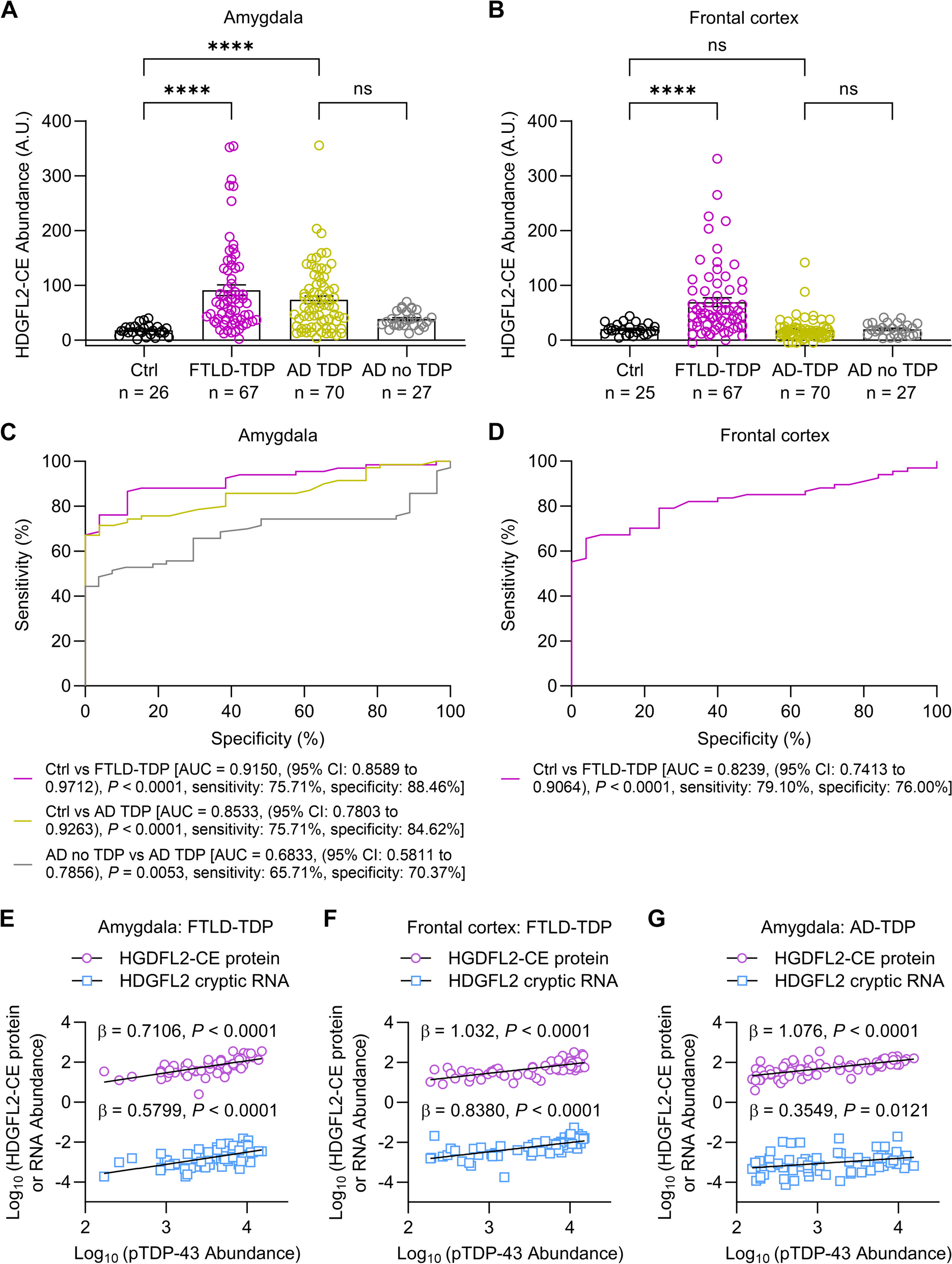
The ATP-binding cassette (ABC) transporter family is a superfamily of highly conserved integral membrane proteins responsible for the transport of various substrates across cellular membranes. Based on amino acid sequence similarity and phylogeny, seven subfamilies from ABCA to ABCG are defined, which classify all 48 functional human ABC transporters [16]. ABC transporters share a characteristic architecture, consisting of four core domains: two nucleotide binding domains (NBD) and two transmembrane domains (TMD) (Fig. 1). The NBDs provide the energy for substrate transport by ATP-binding and ATP-hydrolysis and contain three highly conserved motifs: Walker A and B motifs and a signature (C) motif. The TMDs typically contain six membrane-spanning α-helices and provide a pathway across the membrane for substrate transport [17]. These domains also harbor ligand-binding sites that determine the substrate specificity [18].
Fig. 1
Overview of genetic variants and functional mechanisms involved in the etiopathology of ABCA-transporters ABCA1, 2, 5 and 7 to Alzheimer’s disease. Other diseases in which the described ABCA transporters are implicated are also shown
The ABCA subfamily comprises 12 functional transporters, ABCA1 to ABCA13, with ABCA11 representing a transcribed pseudogene. The A-subfamily is characterized by two large extracellular loops between the first and second helix of each transmembrane domain, which can function as ligand binding sites (Fig. 1) [19]. Several members have been identified as lipid transporters in different body locations [20]. The subfamily can be divided in two subgroups, based on phylogenetic analysis and chromosomal location [17]. The first subgroup of five genes (ABCA5-6 and ABCA8-10) is organized in a head-to-tail cluster on chromosome 17q24, while the second group of seven genes (ABCA1-4, ABCA7 and ABCA12-13) is dispersed on six chromosomes [17].
Over the past years, A-subclass ABC proteins have gained a lot of attention due to their implication in human diseases. To date, mutations in five ABCA genes are causatively linked to monogenic recessive disorders: ABCA1 (Tangier disease), ABCA3 (neonatal surfactant deficiency), ABCA4 (Stargardt disease), ABCA12 (harlequin ichthyosis) and most recently ABCA5 was linked to congenital generalized hypertrichosis terminalis [21,22,23,24,25]. Additionally, rare coding variants in ABCA13, increase the susceptibility to schizophrenia and bipolar disorder [26, 27]. Moreover, GWAS recognized ABCA7 and more recently ABCA1 as risk genes for LOAD [15, 28]. Post-GWAS genetic studies identified common and rare ABCA7 variants that influence AD risk, establishing ABCA7 as an important AD risk gene. Although the underlying mechanism linking ABCA7 risk variants to AD pathogenesis is poorly understood, ABCA7 is functionally involved in several molecular processes linked to AD etiology.
Besides ABCA1 and ABCA7, two additional ABCA members, ABCA2 and ABCA5, have been genetically and/or functionally linked to AD, supporting a broader function of this protein subfamily to the etiopathogenesis of AD (Fig. 1). Interestingly, these ABCA transporters are all implicated in cholesterol homeostasis, a pathway for which an important role in AD has been suggested, as highlighted below. In this review, we will first discuss the link between cholesterol metabolism and AD, before reviewing the genetic and functional evidence linking ABCA1, ABCA2, ABCA5 and ABCA7 to AD.
The link between cholesterol homeostasis and ADCholesterol is a key component of mammalian cell membranes and it is involved in a large number of cellular processes [29]. Membrane cholesterol regulates membrane fluidity, rigidity and permeability by interacting with surrounding bilayer lipids and regulates signal transduction by interacting with transmembrane proteins [29]. The brain contains the highest cholesterol levels in the body, and tight regulation of its synthesis, storage, transport and removal is essential for neuronal functioning [30]. Brain cholesterol mainly originates from de novo synthesis, since systemic lipoprotein uptake is prevented by the blood–brain barrier (BBB) [31]. In the adult brain, cholesterol synthesis is mostly dedicated to astrocytes, which then redistribute cholesterol to neurons, a process mediated by ABCA1 [32]. ABCA1 exports excess cellular cholesterol and phospholipids to apolipoproteins [33]. While apolipoprotein A1 (ApoA1) is the major component of high-density lipoprotein (HDL) particles in the plasma, shuttling cholesterol to the liver for excretion, ApoE is the main cholesterol transporter in the central nervous system and is predominantly produced by astrocytes [33]. In the brain, the HDL-like ApoE-cholesterol-phospholipid complexes can be internalized by neurons, by binding to cell surface receptors, such as the low-density lipoprotein (LDL) receptor [34]. Excess cholesterol can be excreted by conversion to 24-S-hydroxycholesterol, which can readily pass the BBB to be further metabolized by the liver or can be esterified and stored intracellularly as lipid droplets [35, 36]. In addition, it is hypothesized that brain cholesterol is eliminated through the BBB by efflux transporters, such as ABC transporters [37].
β- and γ-secretases mainly operate in cholesterol-enriched membrane microdomains termed lipid rafts, while α-secretase mainly localizes to non-raft regions. High plasma membrane cholesterol levels facilitate the colocalization of APP with β- and γ-secretases, promoting amyloidogenic APP processing and therefore Aβ production [38]. In line, cholesterol depletion promotes the nonamyloidogenic α-secretase cleavage of APP, leading to a reduced Aβ production [39]. Despite the separation of brain and peripheral cholesterol pools, epidemiological studies identified a link between high serum cholesterol levels and AD risk [40]. In parallel, the use of cholesterol-lowering agents, i.e. statins, is associated with lower AD risk [41]. The flux of plasma oxysterols towards the central nervous system following hypercholesterolemia, together with disruption of the BBB might explain the link between serum cholesterol and AD [42].
A first genetic link between AD and lipid metabolism was established when the ε4 allele of APOE was identified as a major genetic risk factor for AD and cerebral amyloid angiopathy (CAA) [43, 44]. ApoE ε4 is suggested to increase Aβ aggregation and decrease Aβ clearance [45, 46]. Indeed, ApoE colocalizes with senile plaques, neurofibrillary tangles, and vascular amyloid [12], and was found to bind Aβ, although the ApoE ε4 isoform shows a decreased Aβ binding affinity [47, 48]. In addition, an isoform-dependent difference in cellular cholesterol efflux is observed, with ApoE ε4 showing the least efflux capacity [49]. Decades after the identification of APOE ε4 as a strong AD risk factor, GWAS in LOAD cohorts identified a high number of risk genes that are implicated in lipid metabolism, including two genes of the ABCA subfamily: ABCA1 and ABCA7 (Fig. 1) [14, 50, 51].
ABCA1In the periphery, ABCA1 promotes the release of cellular cholesterol and phospholipids to lipid-poor apolipoproteins, mainly ApoA1, to generate HDL [52]. Since cholesterol is mainly catabolized in the liver, efflux of excessive cellular cholesterol by ABCA1 to ApoA1 plays a key role in the reverse cholesterol transport pathway in order to deliver HDL to the liver for excretion [53]. The identification of ABCA1 loss of function mutations in patients with HDL-deficiency syndromes, including Tangier disease, confirmed the role of ABCA1 in cellular cholesterol homeostasis [54]. Tangier disease is a recessive disorder characterized by extremely low plasma HDL and ApoA1 levels, intracellular cholesterol depositions, premature atherosclerosis and peripheral neuropathy [54]. The role of ABCA1 in the periphery has been extensively studied. Nevertheless, ABCA1 is highly expressed in the human brain, with the highest expression in neurons and microglia [55]. Studies in mice showed that in the central nervous system, ABCA1 is directly involved in brain cholesterol homeostasis by exporting cholesterol through the BBB [56]. In addition, loss of Abca1 results in a major decrease in ApoE protein levels and ApoE lipidation, as well as an impaired hippocampal neurite morphology in mice, suggesting a role for ABCA1 in AD [33, 57]. Lipidation of ApoE is required for its functioning, including the ability to bind Aβ [48], and a lower lipidation status has been observed in ApoE ε4 compared to ApoE ε3 produced by human iPSC-derived astrocytes [58]. Furthermore, Abca1 deficiency increases Aβ deposition as well as CAA in two AD mouse models [59, 60], and is linked to cognitive deficits in mice [57, 61]. Fitz et al. demonstrate that Abca1 deficiency in an AD mouse model negatively impacts amyloid deposition, Aβ clearance and memory in mice expressing human APOE ε4 but not APOE ε3, suggesting an interaction between ABCA1 and other genetic risk factors [62]. In parallel, overexpression of Abca1 in an AD mouse model reduced fibrillogenesis and deposition of Aβ in the brain, possibly related to the increased lipidation of ApoE [63]. Selective stimulation of Abca1 with an ABCA1 agonist in mice expressing human APOE ε4, increased lipidation of ApoE ε4 and ameliorated ApoE ε4-driven cognitive impairments and brain pathology, rendering it to a similar level as the mice expressing ApoE ε3 [64]. In addition, ABCA1 membrane expression in mice primary astrocytes is diminished in cells expressing human ApoE ε4 compared to ApoE ε3 expressing cells due to a reduced ABCA1 recycling [65]. In parallel, a reduction in ABCA1 protein levels is observed in human astrocytes expressing ApoE ε4, possibly contributing to the ineffective cholesterol efflux in ApoE ε4 cells [66]. Upregulation of ABCA1 and the subsequent increase in APOE lipidation might present a potential therapeutic strategy to ameliorate AD-pathology driven by APOE ε4.
ABCA1 is transcriptionally regulated by oxysterol-activated liver X receptors (LXRs), nuclear receptors which bind to DNA sequences of their target genes as heterodimers with retinoid X receptors (RXRs) to activate transcription [67]. Numerous studies have pursued the use of LXR or RXR agonists to reduce AD-related brain pathology and cognitive impairment, as recently reviewed by Fitz and colleagues [68]. Following LXR activation, a decrease in amyloidogenic APP processing and Aβ secretion has been demonstrated in vitro and in AD mouse models, and improvement of cognitive deficits has been observed in AD mice [69, 70]. These changes were associated with an increased ABCA1 expression and propose the induction of functional ABCA1 as a promising therapeutic option for AD [69, 70].
In vitro experiments with skin fibroblasts derived from two Tangier disease patients carrying homozygous ABCA1 premature termination codon (PTC) or missense mutations leading to a loss of functional protein show an increased production of Aβ compared to control cells [71]. Interestingly, upregulation of ABCA1 expression via a synthetic LXR ligand led to a further Aβ increase in cells carrying a missense mutation (N935S) and stayed the same in cells carrying a nonsense mutation, signaling that functional and full-length ABCA1 is required to benefit from the effect of LXR/RXR agonists on Aβ secretion [71]. This is in line with the clinical phenotype of the N935S patient, who had extremely low HDL levels and developed severe dementia and amyloid depositions by the age of 60 [71]. Another case with a relevant link with AD is a patient carrying a compound heterozygous mutation (D1099Y and F2009S) in ABCA1, who presented with low HDL but no cardiovascular disease, and later developed and died of CAA [72].
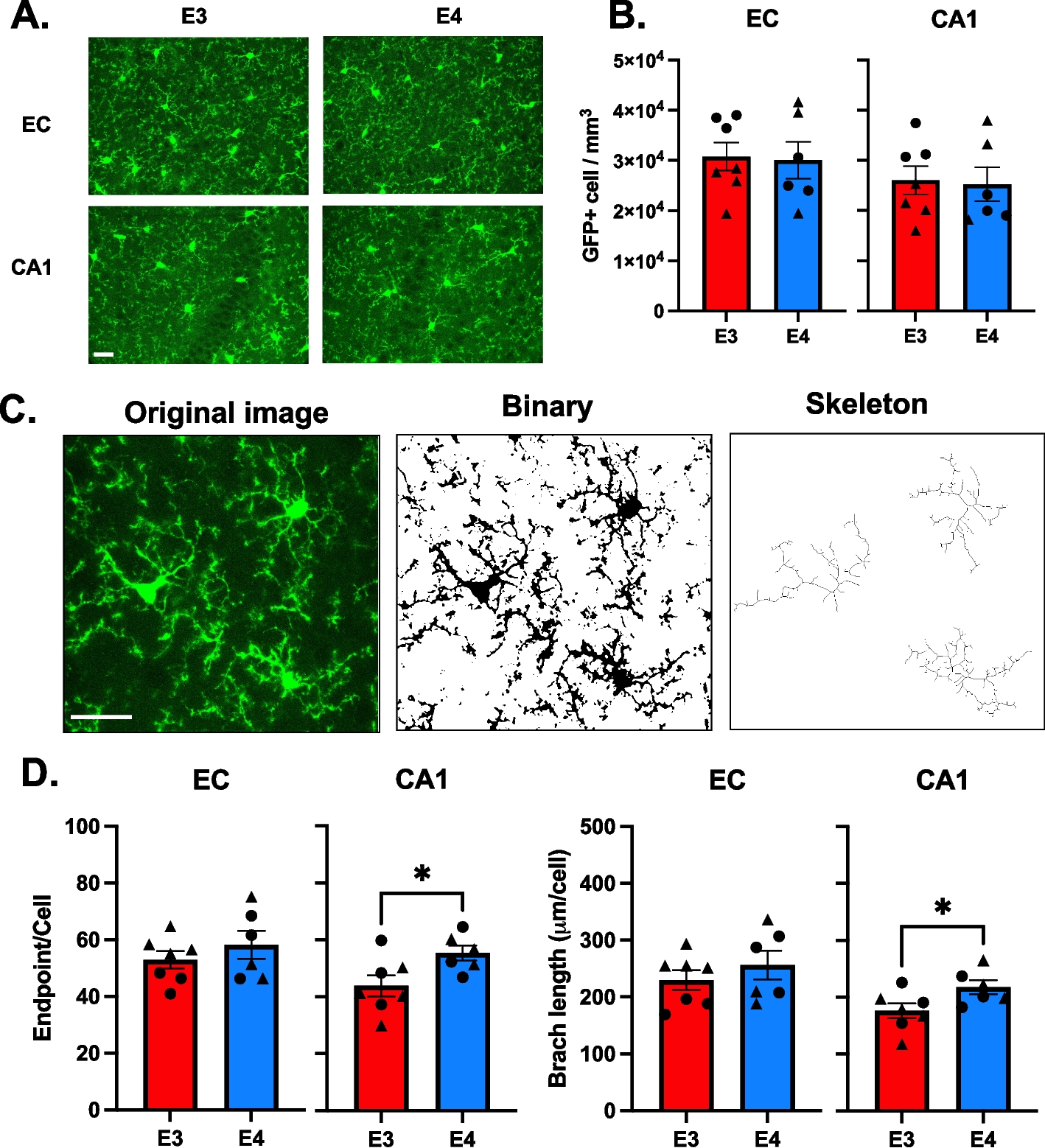
The ABCA1 gene is located near a linkage peak on chromosome 9, previously identified through genome-wide AD linkage studies, and is a good candidate gene given its function in cholesterol homeostasis [73, 74]. Since the early 2000’s, 20 studies exploring the association of ABCA1 common single nucleotide polymorphisms (SNPs) with AD have been published, reporting conflicting results (PubMed, accessed 20 September 2021). An established ABCA1 loss-of-function mutation involved in familial HDL-deficiency, N1800H, is associated with low ApoE plasma levels and a higher risk for AD and cerebrovascular disease [75]. In a family with 4 AD patients, co-segregation of a missense variant (rs137854495; p.A937V) with AD was reported [76]. This variant was previously identified in Tangier disease patients as part of a compound heterozygous mutation [25]. The mutation resides in the Walker A motif of the first NBD and abolishes cholesterol efflux [77]. Interestingly, the same conserved Alanine to Valine substitution in ABCA7 (p.A845V) was identified in a patient with AD (Fig. 2). Subcellular localization studies found that this variant leads to a loss of functional ABCA7 by means of mislocalization from the plasma membrane to the ER [78]. Finally, the largest AD GWAS/GWAX to date, i.e., including AD-by-proxy cases based on parental history of AD, recently identified ABCA1 as a candidate AD gene [14].
Fig. 2
Topological model of ABCA7. Pathogenic ABCA7 missense mutations leading to mislocalization and subsequent loss of functional protein as well as ABCA7 missense mutations corresponding to pathogenic mutations in ABCA transporters implicated in human disease are shown. The ABCA7 sequence was aligned with sequences of ABCA1, 3, 4, 5 and 12. Pathogenic missense mutations in these five genes were downloaded from the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/). ABCA7 missense mutations, previously reported by Le Guennec et al., Sassi et al., Bellenguez et al., De Roeck et al., and Bossaerts et al., that correspond to pathogenic missense mutations in ABCA1, 3, 4, 5 and 12 are shown in the figure [78,79,80,81,82]. Variants marked with a ‘*’ were identified in control individual(s) only. Protein domain and motif information was based on alignment with ABCA1 [83, 84]. ABCA7 missense variants are shown in red. Corresponding pathogenic ABCA1, ABCA4 and ABCA12 mutations are shown in black, blue and purple respectively
ABCA2The ABCA2 gene is located close to ABCA1 on chromosome 9q and encodes a 2436 amino acid polypeptide [85, 86]. ABCA2 mRNA is predominantly expressed in human brain compared to other organs, where it is localized mainly in oligodendrocytes [55, 87]. ABCA2 mRNA expression in macrophages is upregulated in response to cholesterol influx, classifying ABCA2 as a sterol-responsive gene [85]. Given its high expression in the brain, a plausible role for ABCA2 in brain lipid homeostasis is hypothesized [88]. Subcellular localization studies in HEK293 cells overexpressing human ABCA2 show high ABCA2 expression in late endosomes/lysosomes, proposing an intracellular lipid trafficking role, rather than transport across the plasma membrane like ABCA1 and ABCA7 [87]. Human ABCA2 expression in Chinese hamster ovary cells leads to the sequestering of LDL-free cholesterol in the lysosome and blocks its delivery to the endoplasmic reticulum (ER) for esterification, mimicking sterol-deprived conditions, and confirming a role in intracellular cholesterol trafficking [89]. Expression of human ABCA2 in HEK293 cells did not significantly alter cholesterol efflux to ApoA1 or ApoE, which again might reflect the endolysosomal location of ABCA2 [90]. However, later research found a decrease in total and membrane cholesterol levels as well as a reduced cholesterol efflux to ApoE ε3 acceptors in mouse neuroblastoma cells expressing human ABCA2, without perturbing lipid rafts [91]. In addition, ABCA2 regulates cholesterol levels by decreasing LDL receptor mRNA and protein expression [91]. The ABCA2 protein expression dynamics in developing rat brain oligodendrocytes coincide with the myelination process, proposing a role for ABCA2 in myelin formation [92, 93]. In fact, Abca2 knockout mice show abnormal myelin sheet ultrastructure and present with prominent tremor, reduced body weight and hyperactivity, of which the latter two were more prominent in female mice [94]. A second study also observed a tremor in Abca2 knockout mice, and identified alterations in brain sphingolipid levels, but could not confirm abnormal myelin structure [
留言 (0)