
記住我
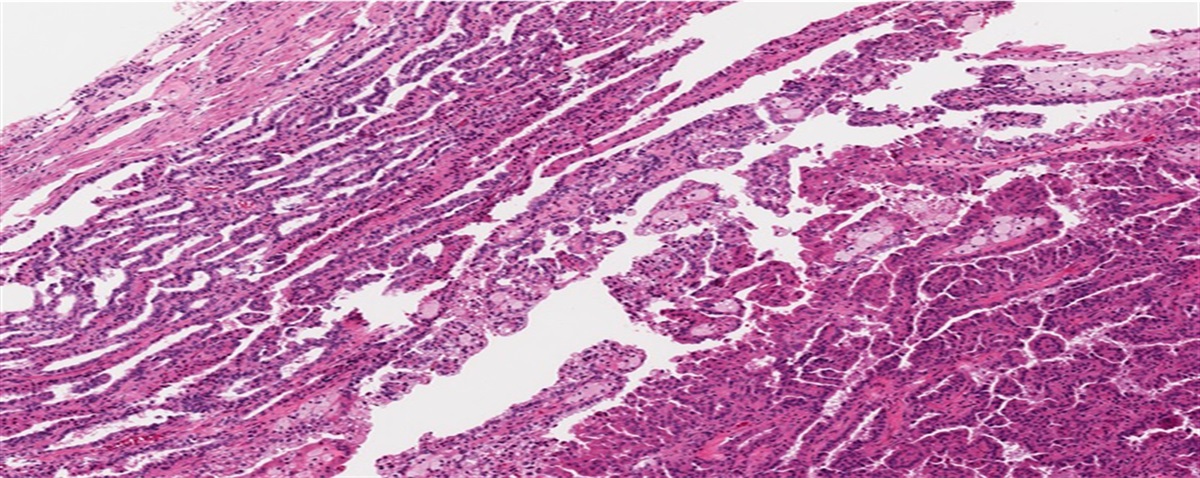
As per the current World Health Organization (WHO) classification, serous tumors are the most common histologic type of tumors of the extrauterine female genital tract.1 Their histologic characterization—that the tumors are composed of fallopian tube or ovarian surface epithelial-like cells—had already been achieved before the first WHO classification.2–5 However, the concept of serous tumors has been changing in accordance with accumulating evidence.1,6,7 The major changes and the nomenclature of serous tumors based on the WHO classification are described in Tables 1 and 2.
 TABLE 1:
TABLE 1: Summary of WHO Classification
 TABLE 2:
TABLE 2: Nomenclature of Serous Tumor in the WHO Classification
The aim of this review is to summarize the accumulated evidence on ovarian serous carcinoma, clarify some unresolved aspects, and speculate on future directions of research. Progress in ovarian serous carcinoma research is summarized in Figure 1. In the following sections, we describe various diagnostic and therapeutic breakthroughs and unsolved problems of serous tumors for 8 dichotomies: high grade versus low grade, ovarian versus extraovarian primary, extrauterine versus uterine primary, sporadic versus hereditary, orthodox versus alternative histology, p53 overexpression versus complete absence of immunophenotype, TP53-mutated versus intact precursor, and therapy responsive versus refractory.
 FIGURE 1:
FIGURE 1: Progress in ovarian serous carcinoma. APST indicates atypical proliferative serous tumor; CLOVAR, CLassification of OVARian cancer; CRS, chemotherapy response score; ESPs, early serous proliferations; FGT, female genital tract; FIGO, International Federation of Gynecology and Obstetrics; FT, fallopian tube; GOG, Gynecologic Oncology Group; HG, high-grade; HGSC, high-grade serous carcinoma; LG, low-grade; LGSC, low-grade serous carcinoma; MP, MicroPapillary variant; NI, noninvasive; PARP, Poly (ADP-Ribose) Polymerase; SBT, serous borderline tumor; SCOUT, Secretory Cell OUTgrowth; SEE-FIM, Sectioning and Extensively Examining the FIMbria; SET, solid, pseudo-endometrioid, and/or transitional cell carcinoma-like; STIC, serous tubal intraepithelial carcinoma; TCGA, The Cancer Genome Atlas; UICC, Union for International Cancer Control.
HIGH-GRADE VERSUS LOW-GRADE SEROUS CARCINOMAConsistently, ovarian tumor classifications have favored a 3-tier grading system of biological behavior: benign, borderline, also known as “carcinoma of low malignant potential,” and carcinoma (Table 2). Such a grading system has been useful for the prediction of the clinical course. Ahead of the first WHO Blue Book, a UICC (Union for International Cancer Control) book of monograph series, entitled “Ovarian Cancer,” had already presented clinical data showing that histologic type and grading of ovarian tumor is closely associated with patient prognosis, and thus emphasized that precise classification of ovarian tumors is required for prediction of their clinical course.4
After the revised WHO classification, further histologic grading of ovarian cancer was required for the prediction of a more accurate prognosis. However, establishment of a histologic grading system was difficult because of various issues, for example, the lack of consensus on whether borderline tumors should be regarded as malignant, methodology of the grading system, and an uninformed format due to the multiple pathologists, institutes, and treatments involved. For instance, FIGO (International Federation of Gynecology and Obstetrics) recommended a 3-tier grading system that was based on architectural findings,8 whereas the WHO classifications entrusted such grading to the observers.5,6
Accordingly, the third WHO classification proposed a 3-tier grading model in ovarian serous carcinoma.9 This grading system was composed of 3 parameters: a predominantly architectural pattern, cytologic atypia, and mitotic counts. Subsequently, Malpica et al10 proposed a 2-tier grading system, which was based on 2 histologic assessments; the principal and supplemental parameters were nuclear atypia and mitotic rate, respectively. As a result, this 2-tier grading system clarified the dualistic nature of serous carcinoma; namely, low-grade serous carcinoma (LGSC) is an indolent malignancy with a borderline component, whereas high-grade serous carcinoma (HGSC) is an aggressive malignant neoplasm without obvious precursor lesions. Thereafter, the Gynecologic Oncology Group (GOG) conducted a comparative study between 2-tiered and 3-tiered grading systems, and favored the binary grading system, which enabled the prediction of clinical outcome simply and precisely.11
The 2 distinctive serous malignant tumors, LGSC and HGSC, were prototypical type I and type II ovarian tumors, respectively.12,13 Type I ovarian carcinoma is a low-grade epithelial malignant tumor, which develops through step-wise tumor progression, also called the adenoma-carcinoma sequence. Type I carcinoma includes 4 of 5 major types of ovarian carcinoma: LGSC, mucinous, endometrioid, and clear cell carcinoma. Significantly, the original concept of type I was based on a unique form of the ovarian tumorigenic pathway mentioned above, not a specific histopathology. Thus, these 4 distinctive tumors were integrated into the type I carcinoma category.
A type I carcinoma, LGSC often intermingles with the benign and borderline serous tumor component and exhibits low cellular proliferation. Interestingly, the micropapillary pattern of serous borderline tumors, which had been already observed at the time of the second WHO classification,14 is considered a hallmark of the distinctive entity, also known as noninvasive LGSC.15 In addition, LGSC and serous borderline tumors have relatively frequent point mutations in the KRAS and BRAF genes.16,17 Originally, these oncogenes had been identified in oncoviruses, and then their orthologues found.18,19KRAS and BRAF participate in RAS-RAF-MEK-ERK signaling,20 and thus the occurrence of these 2 mutational events in low-grade serous carcinogenesis is mutually exclusive.
In contrast, type II ovarian carcinoma is a high-grade epithelial malignant tumor, which almost always harbors a TP53 mutation21,22 and lacks obvious benign and/or borderline tumors,12 except for in rare cases.23 However, the enigmatic precursor of this de novo high-grade carcinoma was finally found to be a microscopic lesion using the SEE-FIM (Sectioning and Extensively Examining the FIMbria) protocol.24 Interestingly, the early form of HGSC, also known as serous tubal intraepithelial carcinoma (STIC), is frequently detected predominantly in the distal fallopian tube and fimbria. Similar to HGSC, STIC shows a high frequency of TP53 mutations.25 Moreover, concurrent STIC and HGSC have identical TP53 mutations and other genetic alternations,26–29 suggesting that TP53 dysfunction in the fallopian tubal cells triggers high-grade serous carcinogenesis.
Epidemiologically, the vast majority of the serous carcinoma cases are diagnosed as high grade,1,12 and therefore, we principally describe HGSC in the following sections.
OVARIAN VERSUS EXTRAOVARIAN PRIMARY HIGH-GRADE SEROUS CARCINOMAOrigin of extrauterine serous carcinoma is one of the most debated topics even in the current practical diagnostic settings. Extrauterine serous carcinoma was simply considered ovarian serous carcinoma until the second WHO classification, when it was modified to include the peritoneum and fallopian tube due to the consolidation of other anatomic sites in the third WHO classification. HGSC is often found in cases of peritoneal carcinomatosis or omental cake.30 Such an advanced serous carcinoma has been sometimes believed to be a primary peritoneal cancer, owing to the concept of the secondary Müllerian system31 or Müllerian neometaplasia.32
In fact, tumors of the fallopian tube were included in the first WHO Blue Book of the female genital tract tumor.33 According to the Blue Book, the majority of fallopian tubal malignant epithelial tumors are adenocarcinomas, which usually arise in the distal part of the fallopian tube, and approximately half are bilateral. The book also mentioned that tubal adenocarcinoma is comparatively rare, and sometimes it is difficult to establish the tubal origin, because of ovarian involvement. Although these simple descriptions sufficiently elucidated the nature of tubal HGSC, the section on fallopian tubal tumor was excluded from the next WHO Blue Book.34
The third WHO Blue Book contained chapters entitled “Tumours of the Ovary and Peritoneum (Chapter 2)” and “Tumours of the Fallopian Tube and Uterine Ligaments (Chapter 3).”7 The former chapter described serous tumors mainly as ovarian-primary and suggested that ovarian surface lining cells are a potent source of ovarian epithelial tumors.35 In contrast, this chapter lacked a detailed description of peritoneal-primary serous carcinoma.36 In contrast, the latter chapter recognized that tubal carcinoma is a rare gynecologic malignancy, and the majority of them are serous carcinomas with high-grade histology. Taken together, either the ovary or the fallopian tube was a possible candidate for being the primary site of extrauterine serous carcinoma, whereas the peritoneum seemed to be an enigmatic one.
To tackle the issue of the origin of extrauterine serous tumors, the latest WHO classification stated the official opinion in the first chapter “Tumours of the Ovary.”1 Ahead of the revision of the WHO classification, the members of the FIGO committee recognized the discrepancies regarding assigning a primary site for HGSC, and therefore they unified a single staging system for the ovary, fallopian tube, and peritoneum.37 Consistent with decision of FIGO, the WHO classification adopted the new staging system and recommended that assignment of the primary site be based on the theory of probable tubal origin. Meanwhile, the ovary and peritoneum are also minor and rare sources of HGSC, respectively, and therefore, primary ovarian and peritoneal HGSC should be considered in cases of advanced pelvic HGSC that lack demonstrable tubal lesions.
Recently, a British and Canadian group proposed a protocol to make the terminology uniform and determine the criteria for tubo-ovarian HGSC classification.38 Consistent with the concept of WHO classification, extrauterine HGSC with STIC or fallopian tube involvement is interpreted as fallopian tube primary, regardless of the tumor burden on the ovary and peritoneum. In contrast, extrauterine HGSC without any tubal intraepithelial and invasive lesion is considered to be primarily ovarian after the confirmation of ovarian involvement. Only pelvic HGSC with no histologically proven adnexal lesions is included in peritoneal primary tumor. This simple diagnostic approach was widely accepted by both clinicians and pathologists, whereas the GOG/WHO 2003 dominant mass criteria39 are still popular only among a minor proportion of gynecologic oncologists.40 In contrast to the tubal origin theory-based diagnostic approach, the GOG criteria indicated HGSC to be ovarian primary,41 suggesting that standardization of the updated criteria is required to reduce interobserver variability in determining the primary site in each HGSC case.
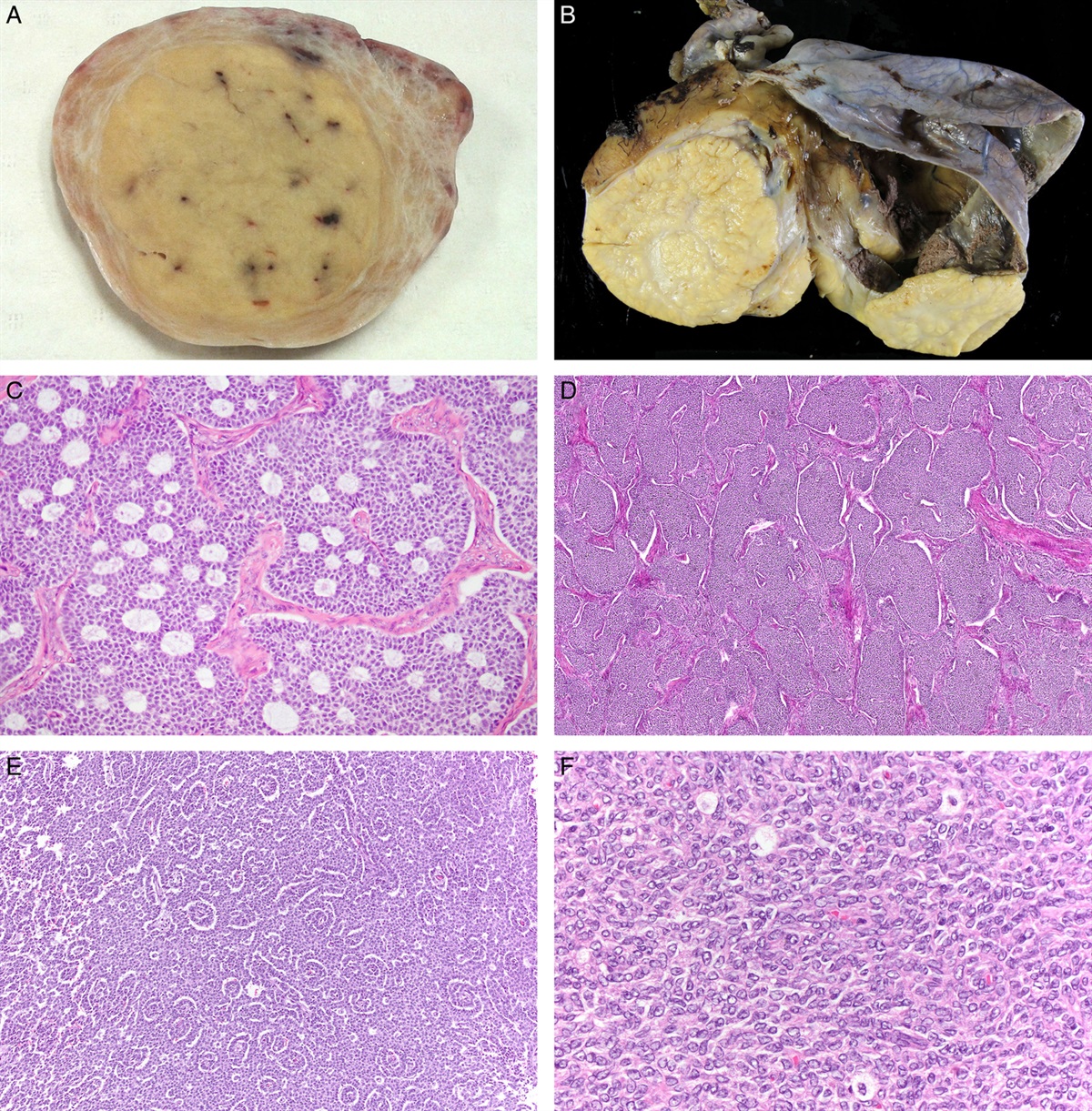
EXTRAUTERINE VERSUS UTERINE PRIMARY SEROUS CARCINOMAApart from being found in the ovary and fallopian tube, serous carcinoma is also found in the uterine corpus and cervix. Unlike the extrauterine serous carcinoma, all other uterine serous carcinomas are considered high-grade malignancies, which most probably harbor a TP53 mutation.42 This relatively minor uterine carcinoma is usually unrelated to endometrial hyperplasia, which is the conventional premalignant endometrial condition, but frequently associated with the noninvasive form, also known as serous endometrial intraepithelial carcinoma (SEIC).43 Interestingly, the causal relationship between endometrial serous carcinoma and SEIC is quite similar to that between high-grade ovarian serous carcinoma and STIC. In contrast, primary uterine cervical serous carcinoma seems to be rare,44 probably due to the precursor lesion being unclear.
Historically, the dichotomy of the tumorigenic pathway in endometrial cancer45 was observed ahead of the ovarian dualistic carcinogenesis model.12 This study,45 spanning 20 years, revealed that the first pathogenic type was low-grade endometrial carcinoma associated with endocrine and/or metabolic disorders, including hyperestrogenism, obesity, and diabetes mellitus. In contrast, the second pathogenic type was high-grade endometrial carcinoma arising from atrophic endometrium without the obvious etiology as seen in the first type. At almost the same time, Lauchlan46 reported the first case series of uterine serous carcinoma which is parallel to ovarian serous carcinoma. As in ovarian serous carcinoma, the prognosis of uterine serous carcinoma was strikingly poor even for noninvasive forms.47 Thereafter, uterine serous carcinoma was added in the second WHO classification of the uterine corpus,34 and gradually the concept of the secondary pathogenic type merged into type II endometrial carcinoma.48
The similarity between endometrial serous carcinoma and extrauterine HGSC poses the question of whether these 2 carcinomas can be distinguished in complex cases, for example, in advanced pelvic serous cancer with SEIC. TP53 mutation analysis of such cases identified 3 mutational patterns: identical TP53 mutations (10/21 cases), discordant TP53 mutations (5/21 cases), and mixed TP53 mutations (6/21 cases).49 The identical cases showed that TP53 mutation in SEIC was compatible to that in extrauterine serous cancer and lacked STIC, indicating that these cases were probably of endometrial origin. In contrast, discordant cases contained the TP53 mutation that was identical to that observed in STIC, not SEIC, and displayed extensive involvement of extrauterine cancer compared with identical cases, suggesting that these cases were likely of tubal origin. Mixed cases, whose metastatic sites contained 2 independent TP53 mutations, were considered synchronous primary endometrial and tubal serous carcinoma.
Another study reported that ~20% (32/161) of uterine serous carcinomas showed tubal involvement, and in approximately half of the cases, tubal involvement was located at the fimbria.50 Practically, immunohistochemical analysis of WT-1, encoded by WT1,51 helps determine the origin of HGSC.52,53 Absent WT-1 is a possible diagnostic clue for endometrial serous carcinoma, but approximately one third of endometrial serous carcinomas are WT-1 positive.54 According to the immunostaining pattern of p53 and WT-1, the tubal lesion of uterine serous carcinoma was considered the most likely metastatic endometrial cancer (26/32 cases).50 In conclusion, to assign a precise primary site to the so-called HGSC, a detailed histologic and genetic examination of the endometrium and fallopian tube is required. Possibly, some of the peritoneal primary serous carcinomas originate from a latent endometrial serous carcinoma, known as the minimal uterine serous carcinoma.55
HEREDITARY VERSUS SPORADIC HIGH-GRADE SEROUS CARCINOMAHereditary cancer syndromes comprise ~10% of all cancer cases and arise from germline mutation of cancer-related genes, whose dysfunction leads to a predisposition to cancer.56 Hereditary breast and ovarian cancers principally arise from BRCA1/2 germline mutation.57–60 The estimated lifetime ovarian cancer risk is 44% and 17% for BRCA1 and BRCA2 mutation carriers, respectively.61 Although there are some uncommon hereditary ovarian cancer syndromes,62 traditional hereditary ovarian cancer is virtually an HGSC. Indeed, the familial ovarian cancer risk is significantly associated with serous and/or moderately to poorly differentiated carcinoma, compared with other major epithelial carcinomas.63 Besides familial history, menopausal hormone therapy is considered the most important risk factor for serous carcinoma.
Hereditary ovarian cancer frequently develops when the BRCA mutation carrier is still fertile. To avoid lethal malignancy, the carriers received risk-reducing salpingo-oophorectomy (RRSO).64 Notably, pathologic assessment revealed that some proportion of the prophylactic surgical specimen had already contained ovarian and tubal carcinoma.65–67 The majority of the detected malignancy was microscopic intraepithelial carcinoma, which tend to be located at the fimbria. This discovery led to the identification of tubal intraepithelial carcinoma, the precursor of HGSC.24
BRCA1/2 dysfunction leads to homologous recombination deficiency (HRD), which results in a condition where the cells are unable to utilize the homologous recombination repair pathway when double strand breaks occur.68 HRD in cancer cells occurs in not only germline BRCA mutation cases but also somatic BRCA mutation or BRCA promotor hypermethylation cases.69 In fact, The Cancer Genome Atlas (TCGA) study revealed that sporadic BRCA1/2 mutation and epigenetically BRCA1 silencing accounts for ~6% and 11.5% of all HGSC cases, respectively.70 Regardless of the type of BRCA dysregulation, cancers with such a phenotype are a potent target for poly (ADP-ribose) polymerase (PARP) inhibitors.71–73 Therefore, genetic and epigenetic investigations would be essential for all the advanced and refractory cancers, and the difference between hereditary and sporadic cancer is reducing with respect to the choice of the optimal molecular therapy.
Although germline BRCA1/2 mutation accounts for ~25% of hereditary breast and ovarian cancers, evidence of other candidate genes involved in inherited cancer has subsequently emerged.74 Interestingly, the 10 candidate genes reported in Walsh et al,75 as well as BRCA1/2, participate in the Fanconi anemia protein-mediated DNA repair system, also called the repair of interstrand crosslinks. According to the National Comprehensive Cancer Network (NCCN) guideline,76 germline BRIP1,77RAD51C,78 and RAD51D79 mutation carriers should take into consideration RRSO of 45- to 50-year olds. This late timing of RRSO, which is typically performed between 35 and 40 years of age, is because the cancer risk caused by these moderate-penetrance cancer susceptible genes is relatively low compared with that caused by high-penetrance genes such as BRCA1/2.80–82
TP53 is also a well-known tumor suppressor gene83 as well as a high-penetrance cancer susceptible gene, which is involved in Li-Fraumeni syndrome.84,85 Germline TP53 mutation predisposes adult females to breast cancer. In contrast, the ovarian cancer risk of germline TP53 mutation is not significant enough to recommend the mutant carrier for prophylactic adnexal surgery despite the prevalence in HGSC.76 This evidence suggests that TP53 mutation is essential in the early phase of high-grade serous carcinogenesis but another driving force is required for it to progress to the malignant form.
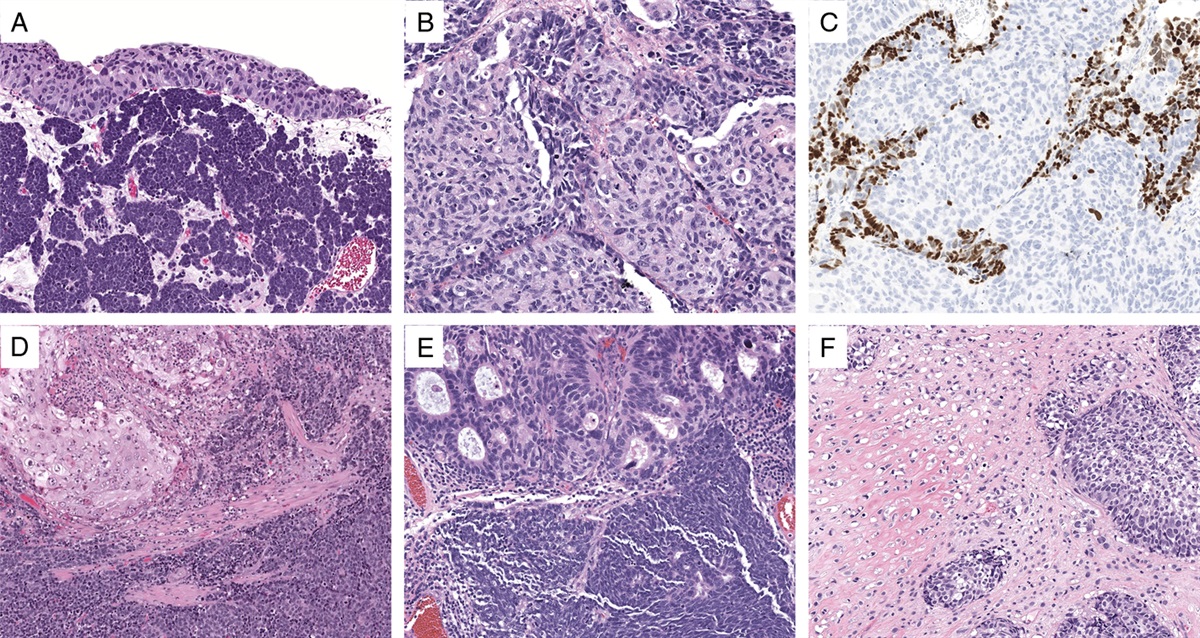
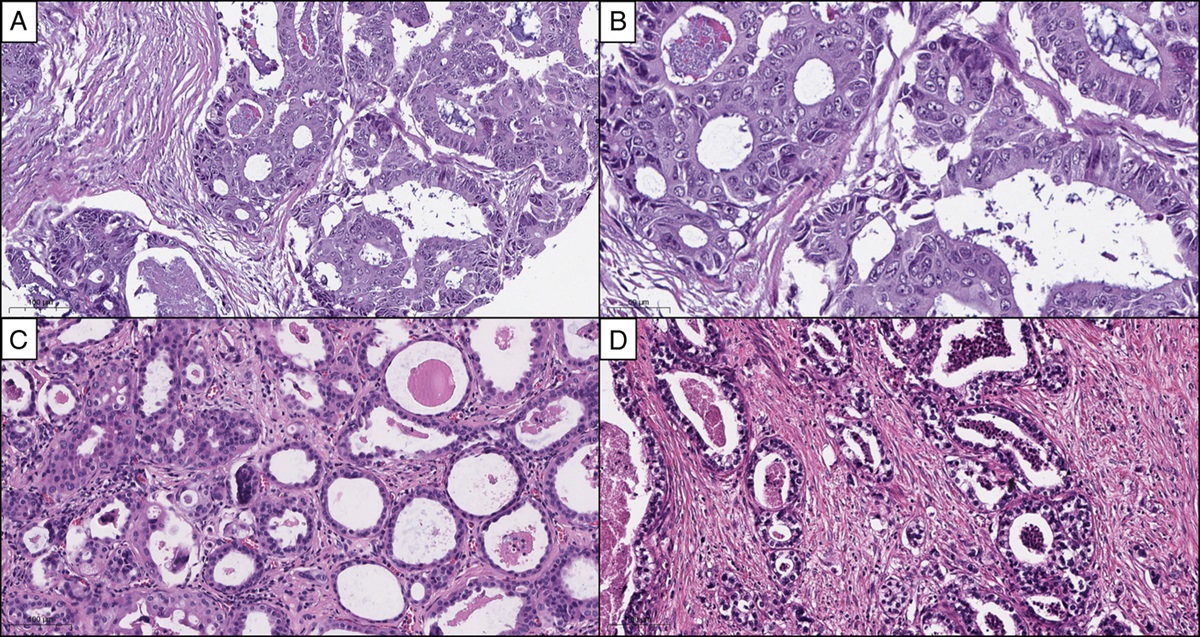
ORTHODOX VERSUS ALTERNATIVE HISTOLOGYThe concept of HGSC has been expanding because of the accumulating evidence of TP53-mutation–related high-grade carcinogenesis. In the latest WHO classification, HGSC consists of the conventional-type and alternative SET-type. Conventional-type HGSC usually shows papillary and slit-like glandular architecture with high-grade nuclear atypia that is greatly deviated from fallopian tubal epithelium, whereas the gene expression profile of serous carcinoma is compatible with that of normal tubal cells.86 Thus, the cellular differentiation in serous carcinoma is confirmable by molecular analysis.
In contrast, the newly added histologic variant, SET-type HGSC predominantly contained solid, pseudo-endometrioid, and/or transitional cell carcinoma–like histologic components.87 Apart from their main histology, TP53 mutation is also detected in the SET variant,88 whereas BRCA dysregulation is frequently associated with the SET variant compared with conventional HGSC.87,89 Surprisingly, the immunoprofile of the SET variant is similar to that of HGSC.90,91 Therefore, it is reasonable to regard this TP53-mutation–related cancer as a category of representative type II carcinoma.
Historically, ovarian transitional cell carcinoma was a prototype of the SET variant. Austin and Norris92 reported that transitional cell carcinoma without Brenner component is more aggressive compared with malignant Brenner tumor with benign Brenner component. This dichotomy in ovarian transitional cell tumor seems to be analogous to the dualistic ovarian carcinoma model.12 A detailed case study revealed that ovarian transitional cell carcinoma lacks Brenner tumor component, but often contains serous, endometrioid, undifferentiated, or unclassified carcinoma.93 Such varied morphology in transitional cell carcinoma suits the name of SET-type HGSC.
Interestingly, HGSC rarely masquerades as mucin-producing carcinoma. According to a major ovarian carcinoma study in Canada,94 ~3% (2/61) of mucinous carcinomas, diagnosed by histologic findings solely, was corrected as HGSC after additional molecular diagnostics. Importantly, mucinous differentiation in transitional cell carcinoma has been reported in the literature.93 To avoid the misinterpretation of such mucinous differentiation, uncommon histology of HGSC should be summarized as STEM (solid, transitional, endometrioid, and mucinous).95
TUMORS WITH p53 OVEREXPRESSION VERSUS p53 COMPLETELY ABSENT IMMUNOPHENOTYPEAberrant p53 expression detected by immunohistochemistry has been known to be a surrogate marker of TP53 mutation in ovarian serous carcinoma.96 Aberrant p53 expression is mainly divided into 2 distinctive patterns: diffuse positive (overexpression)97 and diffuse negative (complete absence).98 The diffuse p53 positivity and negativity are believed to be associated with gain-of-function and loss-of-function p53, respectively.99 Interestingly, the pattern of aberrant p53 expression is closely related to the specific type of TP53 mutation. Most of the p53 overexpression is linked to TP53 missense mutation, which is located at the DNA binding domain. In contrast, complete p53 absence tends to arise from insertion and/or deletion of the TP53 gene. Therefore, the kind of aberrant p53 expression is also a precise predictor of the type of TP53 mutation.
Recently, other p53 immunohistochemical types arising from TP53 mutations have been reported.100 The third aberrant immunophenotype, cytoplasmic p53 expression results from a TP53 mutation that is located at nuclear localized domains. This rare immunophenotype reflects the loss-of-function p53, which is unable to enter the nucleus. Conversely, the wild-type p53 expression pattern is rarely observed with a TP53 mutation due to truncated or 3′ splicing mutations. Therefore, TP53 mutation analysis is needed in complex cases of HGSC with an atypical p53 expression pattern.
TP53-MUTATED VERSUS INTACT PRECURSORTo date, 2 kinds of the minimal precursor of HGSC have been proposed. Lee et al25 was the first to name at least 12 consecutive p53 positive normally appearing tubal epithelial cells the “p53 signature.” Naturally, aberrant p53 expression was predominantly found in the distal fallopian tube and fimbria associated with TP53 mutation. Interestingly, these aberrant cells showed an exclusive secretory phenotype. Although the p53 signature was frequently found in the fallopian tube with STIC, this minimal lesion was also detected in both the healthy and BRCA-mutated fallopian tube. Consequently, the p53 signature is a latent precursor of STIC because TP53 mutation is the main driving force in high-grade serous carcinogenesis, but it does not always seem to develop into STIC.
A major flaw of this representative TP53-mutated precursor, the p53 signature does not include another representative aberrant p53 immunophenotype, the complete absence p53. To detect a null-type p53 signature is a diagnostic challenge because of the benign-like morphology and minimal molecular alteration. Given that p53 dysregulation fails to guard the genome in the tubal cells, the null-type p53 signature maybe detected by a well-established DNA damage marker like γ-H2AX.101,102
Such a concept of “γ-H2AX responsive foci” overlaps with the recently proposed early serous proliferations (ESPs) in the fallopian tube.103 ESPs consist of low-proliferative aberrant p53-expressing cells of overexpression-type and null-type. Interestingly, TP53 mutation analysis revealed that majority of ESPs contained mutations identical to those of concurrent STIC-negative HGSC. In other words, there is the possibility that TP53-dysregulated tubal cells directly migrate to the other site and transform high-grade malignant cells there. This phenomenon, named “precursor escape,” further supported the tubal origin theory in high-grade serous carcinogenesis.
In contrast, secretory cell outgrowth (SCOUT) appeared as a representative intact TP53 precursor.104 The original definition of SCOUT was, at least 30 distinctive secretory epithelial cells with BCL2 expression and without p53 expression, which were different from the background tubal epithelium that consisted of a mixture of secretory and ciliated cells. Unlike the distribution of the p53 signature, SCOUT was found at both proximal and distal ends of the fallopian tube. However, downregulation of PAX2, which is a key event in high-grade serous carcinogenesis,105 was detected in both p53 signature and SCOUT.
These 2 distinct candidate precursors of STIC suggested that high-grade serous carcinogenesis also follows a step-wise progression like type I carcinoma and may consist of multiple pathways depending on the TP53 mutation status. Although the diagnostic approach of tubal intraepithelial lesions was summarized by several groups,106–108 extensive efforts have been made to search for the minimal precursor of HGSC in routine diagnostic setting. Understanding of these microscopic lesions would shed light on the multistep progression of high-grade serous carcinogenesis.
THERAPY RESPONSIVE VERSUS REFRACTORY HIGH-GRADE SEROUS CARCINOMAAs most extrauterine HGSC is found at an advanced clinical stage, neoadjuvant chemotherapy is often administered in these cases. Therefore, the following surgical specimens need histologic assessment of the therapeutic response to make the next clinical decision. Bohm et al109 developed a reproducible histopathologic scoring system, named chemotherapy response score (CRS). In this 3-tiered scoring system, CRS 3, defined by lack of residual tumor cells or minimal neoplastic foci up to 2 mm, is a better prognosis than CRS 1 and CRS 2. In addition, CRS 3-scored patients showed significantly low frequency of postchemotherapeutic elevation of an ovarian tumor marker, CA-125, whereas CA-125 response to neoadjuvant chemotherapy is unable to predict CRS. Collectively, CRS 3 seems to be one of the therapy responsive phenotypes, but its nature remains unclear because of cancer cells vanishing.
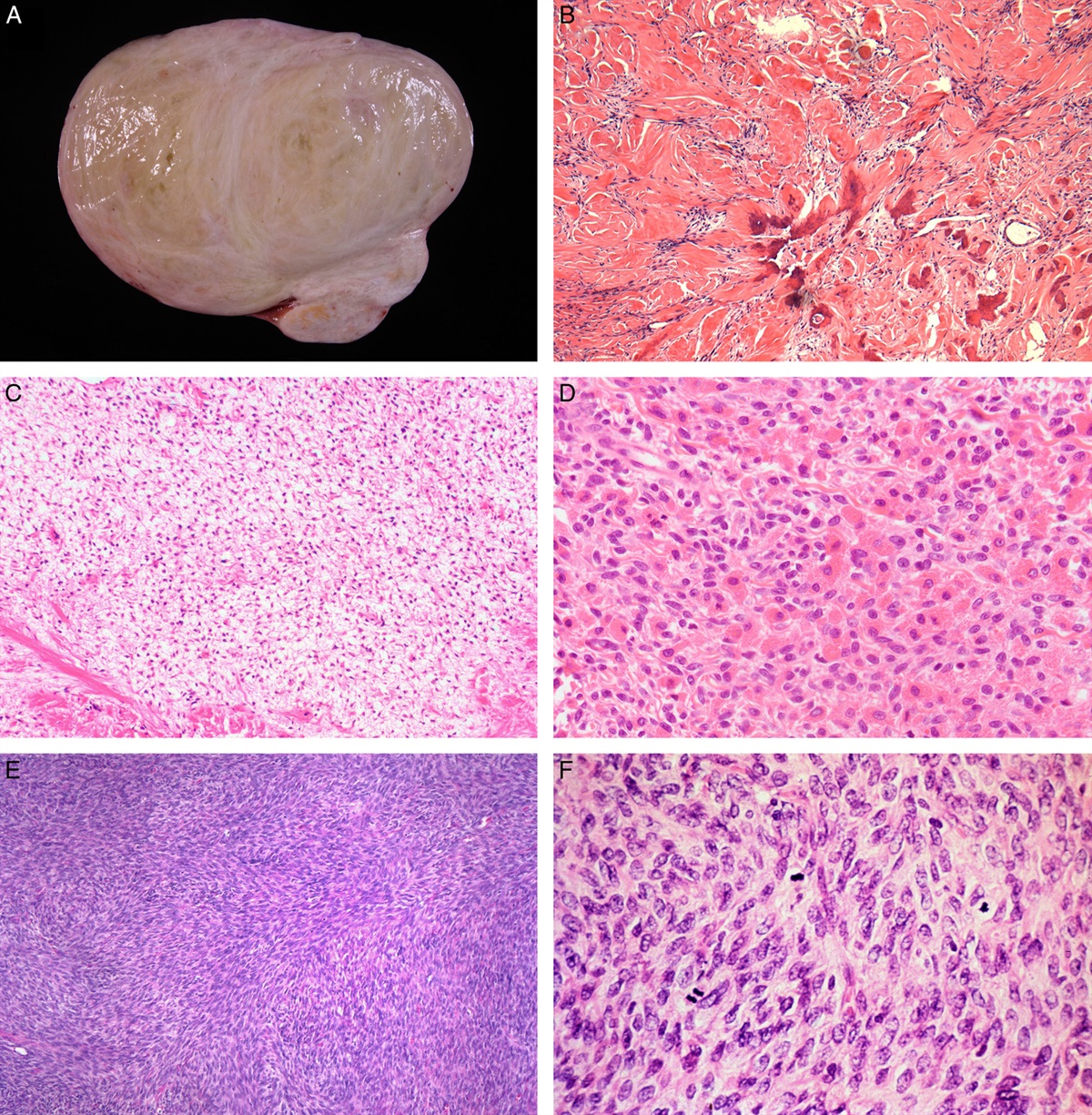
Another therapy responsive phenotype is SET-type HGSC. Although ovarian transitional cell carcinoma was regarded as a high-grade malignancy,92 ovarian carcinoma with predominantly transitional cell pattern (>50%) has a better prognosis compared with ovarian carcinoma with limited transitional cell carcinoma component (<50%).93 Consistent with the finding, better prognosis of SET-type HGSC was reported by another group later.89 Notably, the SET variant is closely associated with 2 unique features: BRCA1 mutation and tumor-infiltrating T lymphocytes. As described above, BRCA1 mutation is associated with HRD, suggesting that such a molecular feature is frail in platinum-based combined chemotherapy because of DNA repair system dysregulation. In addition, tumor-infiltrating T lymphocytes, which are believed to mount an antitumor immune response, had been acknowledged as a good prognostic factor.87,110,111 Taken together, distinctive favorable HGSC is warranted by the trinity of SET-type morphology, BRCA dysregulation, and tumor-infiltrating T lymphocytes.
In contrast, therapy refractory features in HGSC were studied by whole-genome sequencing.112 This study confirmed 2 notable refractory features: CCNE1 amplification and reversion of BRCA1/2 mutation.
The CCNE1 amplification in ovarian carcinoma was first highlighted by a GOG study.113 The patients with high expression of cyclin E1, encoded by CCNE1, showed significantly shorter survival time, and the high expression was associated with CCNE1 amplification. In cell cycle, cyclin E1 acts as a cardinal regulator of G1/S transition, and its overexpression leads to increased cell proliferation through the blockage of the retinoblastoma tumor suppression pathway.114 Consistent with the initial report, several groups described that the CCNE1 amplification or cyclin E1 overexpression is associated with unfavorable prognosis and therapy resistance.115,116 Interestingly, CCNE1 amplification is found in ~20% of HGSCs and for the most part is exclusive of BRCA1/2 dysregulation.112 Furthermore, the CCNE1 locus is on chromosome 19, which usually includes localized and clustered breakpoints in the case of the genome of HGSC cells. These findings indicated that CCNE1 amplification is one of the essential molecular events in high-grade serous carcinogenesis.
Reversion of BRCA1/2 mutation is a unique genetic event that leads to therapy resistance in cancer cells. Reversion of BRCA1/2 mutation stands for the secondary mutation that allows BRCA1/2-mutated cancer cells to restore to wild-type or nearly intact BRCA1/2 state,117–119 allowing these cancer cells to reacquire normal BRCA1/2 function, and resistance to chemotherapy.120 Beside these genetic alterations, demethylation of the BRCA1 promotor region also restores BRCA1 expression and results in chemoresistance.112 Collectively, the BRCA1/2 mutation plays an important role in the early stage of high-grade serous carcinogenesis to form specific genomic patterns effectively.121 Meanwhile, this mutagenic phenotype, derived from BRCA1/2 mutation, is impeditive for cancer cell survival because of unavailability of the DNA repair system. Therefore, reversion of the BRCA1/2 mutation is a reasonable genetic event in the late stage of high-grade serous carcinogenesis.
MOLECULAR CLASSIFICATION OF HIGH-GRADE SEROUS CARCINOMAOwing to emerging evidence of inter-heterogeneity of cancer even in the same histologic-type, one of the current biggest challenges in ovarian cancer research is to classify the molecular phenotype of HGSC. Such interheterogeneity leads to difficulty in predicting prognosis and therapeutic response. Therefore, the purpose of molecular tumor classification is not only to identify their differentiation and/or origin122 but also to predict their clinical course.123
The TCGA project revealed that the survival gene expression signature enables the prediction of clinical outcome in several HGSC datasets, and HGSC is divided into 4 transcriptional subtypes: differentiated, immunoreactive, mesenchymal, and proliferative.70 Although the original TCGA transcriptional subtypes failed to demonstrate their relationship with prognosis, modified classification methods, de novo classification, and CLassification of OVARian Cancer (CLOVAR), were successful in prediction of overall survival.124,125
Currently, clinicopathologic feature of each molecular subtype in HGSC has been unraveled. Notably, the concept of immunoreactive (C2) and mesenchymal (C1) subtypes was already proposed before the TCGA classification.126 The immunoreactive subtype is a favorable prognostic group characterized by rich tumor-infiltrating T lymphocytes, whereas the mesenchymal subtype is the worst prognostic group that shows a small number of tumor-infiltrating T lymphocytes and extensive desmoplasia.124 In addition, the proliferative subtype is compatible with Tothill’s C5 category126 and falls in the relatively worse prognostic group, which highly expresses cell cycle–related genes, including HMGA2.124,127–129 Differentiated subtype is an intermediate prognostic group that overlaps with Tothill’s C4 category, which also contains the recently proposed better prognostic group, antimesenchymal.130
In the molecular classifications of HGSC tumors, the vast majority of categories are based principally on the gene expression profiles of cancer cells, but Murakami et al131 recently proposed an alternate histology-based tumor classification system. Consistent with the molecular tumor classifications proposed previously, histologic classification consists of 4 tumor types that overlap with the TCGA subtypes: mesenchymal transition-type and mesenchymal subtypes, immune reactive-type and immunoreactive subtype, solid and proliferative-type and proliferative subtype, and papilloglandular-type and differentiated subtype. To the best of our knowledge, this novel classification system is the simplest and most cost-effective system to identify the best (immune reactive) and worst (mesenchymal transition) prognostic groups in routine pathologic examination. Such practical subtyping leads to a better understanding of the heterogeneity in HGSC and to progress in diagnosis and treatment of this highly aggressive cancer.
CONCLUSIONIn this review, we briefly described the history of serous tumors, discussed the 8 kinds of dichotomies in serous carcinomas of the female genital tract, and summarized the current molecular classification. Although in the past, the final diagnosis of ovarian tumor was made solely by morphology, current diagnosis has shifted to a combination of histologic and molecular findings. While the power of histologic pathology in unravelling the mystery of the origin of HGSC is undeniable, in future, molecular findings would rank at the top of an integrated diagnosis system for making a decision.
REFERENCES 1. Kurman RJ, Carcangiu ML, Herrington CS, et al. WHO Classification of Tumours of the Female Reproductive Organs. Lyon: WHO Press; 2014. 2. Schiller W. Concept of a new classification of ovarian tumor. Surg Gynecol Obstet. 1940;70:773–782. 3. Bassis ML. An embryologically derived classification of ovarian tumors. JAMA. 1960;174:1316–1319. 4. Gentile F, Junqueira AC. Ovarian Cancer UICC Monograph Series Volume 11. Heidelberg: Springer-Verlag, Berlin; 1968. 5. Serov SF, Scully RE, Sobin LH. Histological Typing of Ovarian Tumours: International Histological Classification of Tumours No 9. Geneva: WHO; 1973. 6. Scully RE, Sobin LH. Histological Typing of Ovarian Tumours: World Health Organization International Histological Classification of Tumours. Berlin: Springer-Verlag; 1999. 7. Tavassoli FA, Deville P. World Health Organization classification of tumours Pathology and genetics of tumours of the breast and female genital organs. Lyon: IARC Press; 2003. 8. Classification and staging of malignant tumours in the female pelvis. Acta Obstet Gynecol Scand. 1971;50:1–7. 9. Silverberg SG. Histopathologic grading of ovarian carcinoma: a review and proposal. Int J Gynecol Pathol. 2000;19:7–15. 10. Malpica A, Deavers MT, Lu K, et al. Grading ovarian serous carcinoma using a two-tier system. Am J Surg Pathol. 2004;28:496–504. 11. Bodurka DC, Deavers MT, Tian C, et al. Reclassification of serous ovarian carcinoma by a 2-tier system: a Gynecologic Oncology Group Study. Cancer. 2012;118:3087–3094. 12. Shih Ie M, Kurman RJ. Ovarian tumorigenesis: a proposed model based on morphological and molecular genetic analysis. Am J Pathol. 2004;164:1511–1518. 13. Kurman RJ, Shih IM. The dualistic model of ovarian carcinogenesis: revisited, revised, and expanded. Am J Pathol. 2016;186:733–747. 14. Burks RT, Sherman ME, Kurman RJ. Micropapillary serous carcinoma of the ovary. A distinctive low-grade carcinoma related to serous borderline tumors. Am J Surg Pathol. 1996;20:1319–1330. 15. Malpica A, Longacre TA. Prognostic indicators in ovarian serous borderline tumours. Pathology. 2018;50:205–213. 16. Singer G, Oldt R III, Cohen Y, et al. Mutations in BRAF and KRAS characterize the development of low-grade ovarian serous carcinoma. J Natl Cancer Inst. 2003;95:484–486. 17. Jones S, Wang TL, Kurman RJ, et al. Low-grade serous carcinomas of the ovary contain very few point mutations. J Pathol. 2012;226:413–420. 18. Sithanandam G, Kolch W, Duh FM, et al. Complete coding sequence of a human B-raf cDNA and detection of B-raf protein kinase with isozyme specific antibodies. Oncogene. 1990;5:1775–1780. 19. Tsuchida N, Murugan AK, Grieco M. Kirsten Ras* oncogene: significance of its discovery in human cancer research. Oncotarget. 2016;7:46717–46733. 20. Samatar AA, Poulikakos PI. Targeting RAS-ERK signalling in cancer: promises and challenges. Nat Rev Drug Discov. 2014;13:928–942. 21. Ahmed AA, Etemadmoghadam D, Temple J, et al. Driver mutations in TP53 are ubiquitous in high grade serous carcinoma of the ovary. J Pathol. 2010;221:49–56. 22. Vang R, Levine DA, Soslow RA, et al. Molecular alterations of TP53 are a defining feature of ovarian high-grade serous carcinoma: a rereview of cases lacking TP53 mutations in the Cancer Genome Atlas Ovarian Study. Int J Gynecol Pathol. 2016;35:48–55. 23. Ramanayake N, Russell P, Yang V. High grade serous intraepithelial carcinoma arising in a benign ovarian serous cyst—a bridge too far? Pathology. 2018;50:485–489. 24. Medeiros F, Muto MG, Lee Y, et al. The tubal fimbria is a preferred site for early adenocarcinoma in women with familial ovarian cancer syndrome. Am J Surg Pathol. 2006;30:230–236. 25. Lee Y, Miron A, Drapkin R, et al. A candidate precursor to serous carcinoma that originates in the distal fallopian tube. J Pathol. 2007;211:26–35. 26. Kindelberger DW, Lee Y, Miron A, et al. Intraepithelial carcinoma of the fimbria and pelvic serous carcinoma: evidence for a causal relationship. Am J Surg Pathol. 2007;31:161–169. 27. Kuhn E, Meeker A, Wang TL, et al. Shortened telomeres in serous tubal intraepithelial carcinoma: an early event in ovarian high-grade serous carcinogenesis. Am J Surg Pathol. 2010;34:829–836. 28. Kuhn E, Kurman RJ, Vang R, et al. TP53 mutations in serous tubal intraepithelial carcinoma and concurrent pelvic high-grade serous carcinoma—evidence supporting the clonal relationship of the two lesions. J Pathol. 2012;226:421–426. 29. Kuhn E, Wang TL, Doberstein K, et al. CCNE1 amplification and centrosome number abnormal
留言 (0)