
記住我

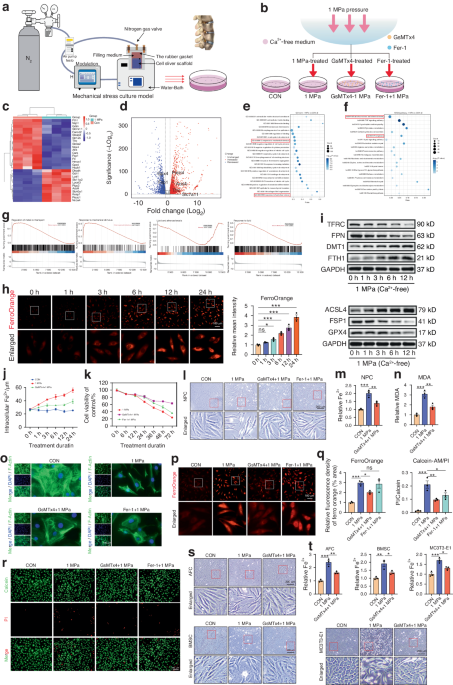
C57BL/6J female mice (9 weeks of age) were purchased from Harlan Israel and housed at the Hebrew University specific pathogen-free facility. tnf-α−/− C57BL6.129S-Tnftm1Gkl/J mice were obtained from The Jackson Laboratory. gfp transgenic C57BL/6-Tg(UBC-GFP)30Scha/J mice were a kind gift from Prof. Reuven Or (Hadassah Medical Center, Jerusalem Israel). Animal use followed protocols approved by the Hebrew University-Hadassah Medical School Institutional Animal Care and Use Committee.
Induction of chronic inflammation by repetitive vaccinationMice were vaccinated with 50 µg heat-killed Mycobacterium tuberculosis H37 Ra (BCG) (Difco Laboratories, Cat#: 231141) once per week over 16 days (Days 0, 7, and 14). The first injection (Day 0) was emulsified in incomplete Freund’s adjuvant (IFA) (Sigma–Aldrich, Cat#: F5506) at a 1:1 ratio (v/v) and administered subcutaneously (s.c.) to the back of the neck. A week later (Day 7), BCG emulsified in IFA was injected into the left footpad (i.f.). After another week (Day 14), a booster immunization containing heat-killed M. tuberculosis without IFA was administered intraperitoneally (i.p.). The mice were sacrificed on Day 16, unless stated otherwise (Fig. 1a). While the second BCG dose (Day 7) was administered to the left footpad, all analyses were performed on the contralateral leg (right) to avoid any effects of local edema around the vaccination site and measure only the systemic effects of chronic inflammation.
DSS-induced colitis – IBDMice were treated with 2.5% (w/v) DSS (Merck-Millipore, cat#: 160110) in the drinking water for 5 days. Weight was monitored for 14 days after DSS treatment, and the mice were sacrificed on Day 19. Colons were removed post-mortem and washed with saline before pictures were taken.
Microcomputed tomography (microCT)Right femora were fixed in PBS containing 4% paraformaldehyde (PFA) for 48 h at room temperature and stored at −20 °C. Bones were scanned using a Skyscan 1174 (Bruker) scanner. The scans were performed with a 7.6-μm voxel size. The mineralized tissues were segmented by a global thresholding procedure. Analyses of trabecular bone were performed in an area that was 0.6 mm below the distal growth plate. The length of the analyzed area of trabecular bone was 3 mm. Analyses of cortical bones were performed on the midshaft region of the bone.
HistomorphometryMice were injected intraperitoneally with calcein (Sigma–Aldrich, Cat#: C0875) on Day 12 and Day 15 of the vaccination protocol to assess bone formation. Undecalcified femora were embedded in Technovit 9100 (Heraeus Kulzer, Cat#: 14655), and 8-µm longitudinal midfrontal plane sections were prepared for dynamic histomorphometric analysis as described before40. Dynamic histomorphometry was performed in the distal trabecular area, 0.6 mm from the growth plate into the femoral shaft. For the determination of bone resorption parameters, tibiae were fixed with 4% PFA for 48 h and decalcified with 10% EDTA for 2 weeks. After decalcification, the bones were embedded in paraffin and sectioned with a Leica microtome to a thickness of 5 μm. The sections were subjected to TRAP staining (Merck-Sigma–Aldrich, Cat#: 387A-1KT) for OC counting and imaged with an Eclipse Ci-L microscope, a DS-U3 imaging system with a DS-Fi2 camera and the NIS elements acquisition program (Nikon) at 20× magnification. For both dynamic and static analyses, ImageJ software was used.
Serum collagen type I crosslinked C-telopeptide (CTX-I) measurementBlood was collected from the heart of terminally anesthetized mice, and the serum was collected after clotting and stored at −80 °C. The CTX-I level was determined using a RatLaps CTX-I detection kit (Immuno-Diagnostic-Systems, Cat#: #AC-06F1) according to the manufacturer’s instructions.
Flow cytometry and antibodiesPeripheral blood, spleen and BM cells were stained with antibodies against CD11b (Pacific Blue, M1/70), Ly6C (FITC, PE, or Alexa Fluor700; HK1.4), Ly6G (APC or biotin, 1A8), Thy1.2 (biotin or APC, 30-H12), B220 (biotin, RA3–6B2), Ter119 (biotin, TER-119), Gr1 (APC, RB6–8C5), CD115 (PE, AFS98), RANK (PE, R12–31), CD117 (PE, ACK2), FLT3 (PE, A2F10), Sca1 (PE/Cy7, D7), MHCII (FITC, M5/114.15.2), CD40 (Alexa Fluor 647, 3/23), CD80 (FITC, 16–10A1), CD86 (PE, PO3), CD11c (PE/Cy7, N418), F4/80 (PE or FITC, BM8), CD120a (APC, 55R-286), CD120b (REA228), CD3ε (APC, 145–2C11), CX3CR1 (PE, SA011F11), TGF-β (APC, TW7–16B4), or CD177 (Alexa Fluor 647, FAB8186R) and isotype controls (RTK2758 and MOPC-173), in combination with unlabeled anti-CD16/32 (clone 93). All antibodies were acquired from BioLegend, except those against MHCII (Tonbo), Sca1 (eBioscience), CD177 (R&D) and CD120b (Miltenyi). All cell surface staining was performed in PBS containing 1.5% fetal bovine serum (FBS) and 0.05% sodium azide (FACS buffer) on ice for 30 min, and then the cells were washed with FACS buffer. In the case of biotinylated antibodies, Alexa Fluor 647-conjugated streptavidin (Jackson) was used after any unbound primary antibodies were washed out. For intracellular staining of CD247, cells were fixed in 1% PFA in PBS for 20 min on ice, permeabilized with 0.1% saponin in PBS for 10 min at room temperature and stained with APC-conjugated anti-CD3ε and FITC-conjugated anti-CD247 (H146)41 in the presence of anti-CD16/32 in FACS buffer containing 0.1% saponin for 30 min on ice. Multicolor flow cytometry was performed using a Galios flow cytometer (Beckman-Coulter), and data were analyzed using FCS express v6 (DeNovo software).
Isolation and sorting of Ly6ChiCD11bhi/lo immature monocytes (iOCPs/hOCPs)Monocytes were isolated from spleens and BM by magnetic separation (StemCell Technologies, Cat#: 19861A). The isolated monocytes were stained for Ly6C and CD11b, and Ly6ChiCD11bhi (iOCPs) and Ly6ChiCD11blo (hOCPs) cells were sorted using a FACSARIA III (BD). The sorted cells were collected in MEM-α (Biological Industries) containing 15% FBS and 1.5% pen-strep solution. The samples were concentrated and counted twice using a TC20 automated cell counter (Bio Rad).
OC culturesSorted cells (104) were cultured in 96-well (flat-bottomed) plates in 200 µL MEM-α containing 10% FBS, 1% pen-strep solution, 20 ng·mL−1 RANKL (PeproTech, Cat#: 315–11–10), and 10% CMG14–12 conditioned medium as the source of M-CSF (equivalent to the bioactivity of 20 ng·mL−1 recombinant M-CSF), prepared as previously described42 (i.e., OC differentiation medium). Cultures were kept at 37 °C and 5% CO2 until TRAP staining. The medium was changed every 3 days. OCs were detected with a TRAP staining kit (Merck-Sigma–Aldrich, Cat#: 387A-1KT) as TRAP+ multinucleated cells (≥3 nuclei). Cultures were imaged using an Eclipse Ci-L microscope, a DS-U3 imaging system with a DS-Fi2 camera and the NIS elements acquisition program (Nikon) at 2× and 10× magnification, and the OC area was measured with Image-Pro Plus V6 (Media Cybernetics).
Ex vivo T-cell suppression assayNinety-six-well (U-shaped) plates were precoated with anti-CD3ε (145–2C11) (3 μg·mL−1) and anti-CD28 (37.51) (3 μg·mL−1) antibodies in 100 µL of 0.1 mol·L−1 borate buffer (pH = 8.5) for 24 h at 4 °C. The wells were then blocked with borate buffer containing 1% FBS for 1 h at room temperature and washed with PBS three times. T cells were isolated from normal spleens by magnetic separation (StemCell Technologies, Cat#: 19851 A) and stained with CFSE (Thermo Fisher, Cat#: C34554) as previously described20. A total of 2 × 105 CFSE-labeled T cells were seeded in RPMI medium (Gibco) supplemented with 8% FBS, 2 mmol·L−1 L-glutamine and 1% pen-strep solution (all from Biological Industries). The cultures were incubated at 37 °C and 5% CO2 for 1 h. Sorted iOCPs and hOCPs were seeded on top of the T cells at different T cell:MDSC ratios, and the cocultures were returned to the incubator for 80 h. Samples were harvested in cold FACS buffer and stained with antibodies against Thy1.2. The proliferation index was determined with an FCS-Express V6 proliferation analyzer (DeNovo software).
Cell cycle stainingSurface staining of 2 × 106 cells with biotinylated anti-Thy1.2, B220, Ly6G and Ter119 antibodies, in combination with anti-Ly6C-PE, anti-CD11b-FITC and anti-CD117-PE/Cy7 antibodies, was performed on ice for 30 min. The cells were stained with streptavidin-Alexa Fluor 647 for 15 min on ice. Then, the cells were fixed and permeabilized as in the CD247 staining protocol and stained with 10 μg·mL−1 DAPI (Merck-Sigma–Aldrich, Cat#: D9542) for 15 min on ice. Analysis was performed with an FCS-Express V6 multicycle DNA analyzer (DeNovo software).
Immune effector differentiation assayA total of 105 sorted cells were cultured in 200 µL MEM-α containing 10% FBS, 1% pen–strep solution and 20 ng·mL−1 GM-CSF (PeproTech, Cat#: 315–03–10) or M-CSF (PeproTech, Cat#: 315–02–10). The cultures were kept at 37 °C and 5% CO2 for 3 days and then harvested by strong pipetting in cold 5 mmol·L−1 EDTA and 5 mmol·L−1 EGTA PBS buffer. Cells were stained for CD11c, MHCII and CD40 to assess DC and macrophage differentiation by flow cytometry.
Pit formation assayA total of 5 × 104 sorted cells were cultured on Osteo assay surface plates (Corning, Cat# #3989) (96-well) in 200 µL of OC differentiation medium and incubated at 37 °C and 5% CO2 for 10 days until the OCs were exhausted in all cultures. The cultures were bleached with 6% sodium hypochlorite solution for 30 min, washed extensively with deionized water and air dried for 2 h. The cultures were imaged using an Eclipse Ci-L microscope, a DS-U3 imaging system with a DS-Fi2 camera and the NIS elements acquisition program (Nikon) at 4× and 20× magnification, and pit area was determined with Image-Pro Plus V6 (Media Cybernetics).
Preparation of pure OC samples for proteome analysisiOCPs and hOCPs were sorted from the BM of inflamed mice. A total of 3 × 105 iOCPs or hOCPs were cultured in four technical replicates in 2 mL of MEM-α supplemented with 10% FBS, 1% pen–strep solution, 20 ng·mL−1 RANKL, and 25 ng·mL−1 recombinant M-CSF (PeproTech, Cat#: 315–02–10) in 12-well plates. The hOCP cultures were kept at 37 °C and 5% CO2 for 3 days, while the iOCP cultures were incubated for 5 days. Cultures were harvested using Accutase (Merck-Sigma–Aldrich, Cat#: B6964) for 40 min at 37 °C, and technical replicates were pooled together. The harvested cells were washed and resuspended in MEM-α supplemented with 10% FBS, 1% pen–strep solution and 10 μg·mL−1 Hoechst 33342 (Merck-Sigma–Aldrich, Cat#: B2261) for 30 min on ice for staining. The cells were washed; resuspended in MEM-α supplemented with 10% FBS, 1% pen–strep solution, 5 mmol·L−1 EDTA and 100 μg·mL−1 DNAse I (Merck-Sigma–Aldrich, Cat#: DN25); and filtered gently through a 100-µm nylon cell strainer. Mature OCs were sorted using an ARIAIII cell sorter (BD) as cells containing three or more nuclei. Sorting was performed at ~2 500 events/s, and cells were collected in MEM-α containing 10% FBS and 1% pen–strep solution. The sorted cells were concentrated, washed four times with 1 mL of ice-cold sterile PBS and pelleted for protein extraction. This protocol was performed as previously reported31.
Lineage tracing of iOCP-OCs and hOCP-OCsControl and inflamed WT mice received 106 iOCPs or hOCPs sorted from the BM of inflamed gfp mice by intratibial injection into the right leg. The inflamed recipients received a single intrafootpad vaccination in the left leg 7 days before the implantation. Recipients were sacrificed 7 days after implantation. The right tibia and femur were fixed in 4% PFA–PBS for 24 h at 4 °C and equilibrated in 15% and 30% sucrose PBS reciprocally, each overnight; no decalcification was performed. Bones were embedded in OCT and frozen at −80 °C. Consecutive 7-µm sections were cut using a Leica cryostat as previously described43. Briefly, 7-µm cryosections were made on CryoJane tape windows (Leica, Cat#: 39475214) and then transferred to carrier slides freshly coated with a thin layer of Norland optical adhesive #63 (Norland, USA) by UV (375 nm) illumination for 30 s. Consecutive sections were alternately stained for TRAP and GFP to detect donor-derived OCs. TRAP staining was performed using a leukocyte phosphatase detection kit (Merck-Sigma–Aldrich, Cat#: 387 A) according to the manufacturer’s instructions. For GFP detection, slides were quenched in 3% H2O2 for 10 min, followed by 1 h of blocking in 5% normal horse serum in CAS-block (Thermo-Fisher, Cat#: 008120) containing 0.05% Tween-20. An anti-GFP antibody (Abcam, Cat#: ab6673) was used at a 1:500 dilution in PBS containing 0.05% Tween-20 and 5% horse serum for 1.5 h. Peroxidase-conjugated horse anti-goat IgG (Vector labs, Cat#: MP-7405) was added for 0.5 h, and detection was performed using DAB (Abcam, Cat#: ab64238). Hematoxylin was used for counterstaining. Mounted samples were imaged with an Olympus microscope and a DP74 camera (Olympus) at 6.3× magnification.
Quantitative PCRTotal RNA was extracted from snap-frozen tibiae by homogenization in Tri-reagent (Sigma–Aldrich). cDNA was prepared with m-MLV reverse transcriptase (Quanta, Cat#: 95047–100). Quantitative real-time PCR (ABI 7900) was performed using SYBR green (Thermo Fisher Scientific, Cat#: 11744500). The primer sequences are listed in Supplementary Table 1, and each primer pair was situated across exon-exon junctions. Murine ubiquitin C was used as a housekeeping gene in all experiments. All results are presented as the relative quantity.
Protein extractionPellets of sorted cells were lysed in 2% SDS in 25 mmol·L−1 Tris (pH = 8) supplemented with 10 mmol·L−1 DTT, 1 mmol·L−1 PMSF and 1 mmol·L−1 EDTA on ice. The lysates were sonicated to 20 kJ three times in ice-cold water. Samples for western blot analyses were mixed with loading dye, incubated for 10 min at 95 °C and stored at −20 °C. Lysates for proteomic analyses were incubated at 50 °C for 10 min and stored at −20 °C.
Mass spectrometry (MS) sample preparationProtein concentrations were determined with Coomassie dot blotting using known BSA standards. Reduction/alkylation was performed for 5 min at 50 °C with 10 mmol·L−1 DTT and for 25 minutes at 45 °C with 40 mmol·L−1 IAA in the dark, and the reaction was quenched with 20 mmol·L−1 DTT at RT.
Next, for OCPs, automated SP3 cleanup was performed as previously described44. In summary, the Bravo system was programmed to process four replicates of the four experimental conditions simultaneously, carrying out all handling steps, including aliquoting of magnetic beads, protein clean-up by SP3, protein digestion, and peptide recovery in a 96-well plate. A starting sample volume of 20 μL containing 7.5 μg of sonicated lysates was combined with 5-μL aliquots of a suspension of washed magnetic beads (50 μg·μL−1). Then, 25 μL of 100% acetonitrile (ACN) was added to each sample, followed by 18 min of incubation off the magnetic rack with cycles of agitation. The sample plate was incubated on the magnetic rack for an additional 5 min. Next, the beads were washed two times with 200 μL of 80% EtOH and one time with 100% ACN. Upon removal of residual washing solvents, the beads were resuspended in 35 μL of 100 mmol·L−1 ammonium bicarbonate and 5 μL of 0.05 μg·μL−1 trypsin. Next, the plate was shaken at 1 500 r·min−1 for 60 s before being transferred to the heating deck position for overnight digestion at 37 °C. Digestion was stopped by manually adding 2 μL of 10% trifluoroacetic acid (TFA). The solution was sonicated in a water bath for 5 min and incubated on a magnetic rack for 2 min. Finally, the peptide-containing supernatants were transferred into a new 96-well plate. Peptide solutions (~2.5 µg, 15 μL) were analyzed as single-shot MS injections, and 5 μg from each sample was combined and fractionated off-line using reversed-phase high-pH fractionation. For OCP-OCs, SP3 cleanup was performed manually. Two microliters of washed SP3 magnetic beads (50 μg·μL−1) was combined with 2 μg of protein lysate in 25 μL. ACN was added to a final concentration of 60%, and the beads were washed twice with 80% EtOH. The proteins were eluted in 18 μL of 50 mmol·L−1 ammonium bicarbonate containing 40 ng of Trypsin/Lys-C mixture and digested for 16 h. The reaction was stopped by the addition of 2 μL of 10% formic acid. One microgram of peptide solution was analyzed as single-shot MS injections in two technical replicates.
ChromatographyFor LCMS analysis, peptides were separated using an Easy NanoLC 1200 fitted with a trapping (Acclaim PepMap C18, 5 μm, 100 Å, 100 μm × 2 cm) and an analytical column (Waters nanoEase MZ Peptide BEH C18, 130 Å, 1.7 µm, 75 µm × 25 cm). Solvent A was 0.1% (v/v) formic acid (FA) in ddH2O, and solvent B was 80% ACN and 0.1% (v/v) FA in ddH2O. Samples were loaded onto the trapping column with a constant flow of solvent A at a maximum pressure of 800 bar. Peptides were eluted at a constant flow of 0.3 μL·min−1 and temperature of 55 °C and maintained using a HotSleevePlus column oven (Analytical Sales and Services). During elution, the percentage of solvent B was increased linearly from 3 to 8% in 4 min, from 8% to 10% in 2 min, from 10% to 32% in 68 min, from 32% to 50% in 12 min, and from 50% to 100% in 1 min. Finally, the gradient was finished with 7 min in solvent B, followed by 11 min at 97% solvent A.
Fractionation of the combined OCPs occurred on an Agilent Infinity 1260 LC system (Agilent) using a Phenomenex Gemini 3 μmol·L−1 C18, 100 × 1 mm column (Phenomonex). Buffer A was 20 mmol·L−1 NH4COOH, and buffer B was 100% can. The following gradient was used: 0–2 min, 0% B; 2–60 min, linear gradient to 65% B; 61–62 min, linear gradient to 85% B; 62–67 min, 85% B; and 67–85 min, 0% B. The eluates were collected in 40 fractions and combined in 16 fractions. The 16 fractions were dried with a SpeedVac, and the peptides were resuspended and cleaned using an Oasis PRiME HLB μElution Plate (Waters) and finally resuspended in 15 μL 0.5% TFA.
Proteomic data acquisitionPeptides were introduced into mass spectrometers via a Pico-Tip Emitter 360 μm OD × 20 μm ID; 10 μm tip (New Objective). The capillary temperature was set at 275 °C.
OCP samples were analyzed on a Q-Exactive HF Orbitrap mass spectrometer (Thermo Fisher Scientific) with a spray voltage of 2 kV. Full-scan MS spectra with a mass range of m/z 350 to 1 500 were acquired with the Orbitrap at a resolution of 60 000 FWHM. The filling time was set to a maximum of 50 ms with an automatic gain control target of 3 × 106 ions. The top 20 most abundant ions per full scan were selected for MS2 acquisition. Dynamic exclusion was set for 25 s. Isotopes, unassigned charges, and charges of 1 and >8 were excluded. For MS2 scans, the normalized collision energy was set to 26, and the resolution was set to 15 000 FWHM with automatic gain control of 1 × 105 ions and a maximum fill time of 50 ms. The isolation window was set to m/z 2, with a fixed first mass of m/z 110.
OCP-OC samples were analyzed on a Fusion Orbitrap mass spectrometer (Thermo Fisher Scientific) with a spray voltage of 2.5 kV. Full-scan MS spectra with a mass range of m/z 375 to 1 500 were acquired with the Orbitrap at a resolution of 60 000 FWHM. The filling time was set to a maximum of 50 ms with an automatic gain control target of 250%. Master scans were performed every 3 s. Ions with intensities above 5 × 103 were selected for MS2 acquisition and detection with an ion trap. Dynamic exclusion was set for 20 s. Isotopes, unassigned charges, and charges of 1 and >4 were excluded. For MS2 scans, the normalized collision energy was set to 33, with standard automatic gain control and a maximum fill time of 50 ms. The isolation window was set to m/z 1.6.
Proteomic data processingRaw files were processed using MaxQuant (version 1.6.2.6)45. Technical replicates were combined using identical sample names. A search was performed against the mouse UniProt database (20180716_Uniprot_mus-musculus_canonical_reviewed) using the Andromeda search engine with the following search criteria: enzyme was set to trypsin/P with up to two missed cleavages. Carbamidomethylation (C) and oxidation (M)/acetylation (protein N-term) were selected as fixed and variable modifications, respectively. The first and second search peptide tolerances were set to 20 and 4.5 ppm, respectively. Protein quantification was performed using the label-free quantification (LFQ) algorithm of MaxQuant. MS2 spectra were not required for the LFQ comparison. In addition, intensity-based absolute quantification (iBAQ) intensities were calculated with log fit enabled. Identification transfer between runs via the matching between runs algorithm was allowed. Peptide and protein hits were filtered at a false discovery rate of 1%, with a minimal peptide length of 7 amino acids. The reversed sequences of the target database were used as a decoy database. All remaining settings were default MaxQuant settings. LFQ values were extracted from the protein groups table and log10-transformed for further analysis. Proteins that were only identified by a modification site, contaminants, and the reversed sequences were removed from the dataset. All further analyses were performed using either Perseus (version 1.6.1.3)46 or R software (version 3.3.3)47 packages available through BioConductor48. In particular, differential expression analysis of the samples was performed using Limma moderated t-statistics (R package version 3.30.1)49. The Benjamini–Hochberg correction was used to calculate adjusted P values.
Only protein groups with a valid measurement in two out of four replicates were subjected to the Limma algorithm. LFQ values that were missing in all replicates for a specific experimental condition were imputed using values obtained from a random distribution having a mean and an SD equal to the minimum LFQ value measured in the experiment. The random distribution was obtained with the urnorm function of the Runuran package (version 0.24) with the lower boundary set to 0.
Principal component (PC) analysis was performed using the prcomp function. GO term enrichment analyses were performed using the enrichGO function of the clusterProfiler package (version 3.2.14). The Benjamini–Hochberg FDR was set at 0.05 for the enrichment assay. For OCPs, the analysis was performed separately for PCs 1–4 on the top or bottom 5% of proteins sorted according to respective PC loadings. For OCP-OCs, either positively or negatively differentially expressed proteins (Limma, Benjamini–Hochberg FDR = 0.05) were used. The entire list of proteins detected in each dataset was used as the background.
All R scripts used in data analyses and figure generation are available upon request.
Western blot analysisOne microgram of total protein was loaded onto a 12% polyacrylamide gel. The proteins were blotted onto a nitrocellulose membrane. For the detection of RAGE and PP2Ac, membranes were blocked with 5% BSA in TBST and probed with an anti-RAGE (Abcam, Cat#: Ab3611) or anti-PP2Ac (R&D, Cat#: AF1653) antibody, followed by incubation with peroxidase-conjugated donkey anti-rabbit IgG (BioLegend, Cat#: 406401). For the detection of S100A8/A9, membranes were blocked in 10% skim milk in PBST and probed with goat polyclonal antibodies against S100A8 (R&D, Cat#: AF3059) and S100A9 (R&D, Cat#: AF2065), followed by incubation with peroxidase-conjugated rabbit anti-goat IgG (Jackson ImmunoResearch Labs, Cat#: 305–035–045). Chemiluminescence detection was performed with Clarity western ECL (Bio-Rad, Cat#: 170–5061), blots were captured with a ChemiDoc XRS + molecular imager (Bio-Rad), and densitometry was evaluated with Image Lab (Bio-Rad).
Statistical analysisAll data are presented as the median (line), 25th–75th percentile (box) and range (whiskers). An exact two-tailed Mann–Whitney test was performed for comparisons of two groups. To compare multiple groups to a single control, an exact Kruskal–Wallis test followed by Dunn’s test was performed. For all quadruple comparisons (i.e., inflamed and control WT/tnf-α−/−, inflamed and control iOCP/hOCP, etc.), the effect of each experimental condition on the four experimental groups was examined separately using an exact Kruskal–Wallis test; those few that did not produce significant results were not analyzed further. All other effects were then tested separately using exact two-tailed Mann–Whitney tests, and multiple comparisons within each experiment were adjusted using the Bonferroni correction. To compare iOCPs to hOCPs, which were dependent variables as they came from the same mouse, an extra penalty and Holm correction were used to adjust for multiple comparisons. P values less than 0.05 were considered significant.
留言 (0)