
記住我


The specialized functions and environments of cellular organelles are highly dependent on their unique protein compositions and how these resident proteomes are maintained. While this is established by elaborate protein targeting mechanisms that guide newly synthesized molecules to the correct organellar destination, the proteomes of individual organelles are unique and dynamic, continuously being shaped and maintained by a variety of quality control systems working in parallel. These systems simultaneously promote the retention of appropriate forms and the clearance of proteins whose folding, assembly, localization, or abundance is incorrect. Thus, protein quality control systems play a central role in organellar and cellular homeostasis. Highlighting their importance, defects in quality control components are often incompatible with life or optimal viability, or result in devastating diseases (Bhattacharya & Qi, 2019; Needham et al, 2019).
As a large, multifunctional organelle, the endoplasmic reticulum (ER) houses robust protein quality control systems, on which it relies heavily. First and foremost, the ER is the main site for membrane protein and secreted protein biogenesis, lipid biosynthesis, and for storage of intracellular calcium. The ER also accommodates proteins and protein complexes required for many other cellular functionalities including innate immune signaling, stress responses, and metabolism. These activities take place throughout a continuous network of sheet-like cisternae and dynamic membrane tubules, defining a single luminal compartment (Westrate et al, 2015). Moreover, the extensive structure of the ER impacts the overall architecture and organization within eukaryotic cells. For example, the nuclear envelope, essential for defining the nucleus and its organization, is formed by apposed ER sheets—the inner and outer nuclear membranes—separated by the ER lumen. Also, ER tubules extending into the cytoplasm contact other organelles such as mitochondria and endosomes, thereby influencing both their positioning and dynamics. Thus, spatially coordinating (and compartmentalizing) these concurrent processes requires the composition of various ER domains to be continuously surveyed by protein quality control systems.
The best-characterized protein quality control system of the ER is the ubiquitin-proteasome-dependent process of ER-associated protein degradation (ERAD). While extensively linked to the degradation of unwanted byproducts of protein biogenesis, such as misfolded and unassembled/orphaned proteins, it is now clear that ERAD and the ubiquitination machineries on which it relies also play important roles in continuously shaping the established ER proteome, and consequently its multiple functions. By promoting the regulated degradation of various ER-localized proteins, ERAD exerts an aspect of post-translational control over lipid homeostasis, nuclear envelope organization, inter-organelle contact (e.g., mitochondria, endosomes), calcium flux, and even innate immune signaling. Autophagy-dependent and -independent clearance through endo-lysosomes (Fregno & Molinari, 2019; Molinari, 2021), or direct extraction of mislocalized membrane proteins (McKenna et al, 2020; Dederer & Lemberg, 2021), are alternative strategies for regulating ER composition. Here, we will however focus on the recent advances in understanding the fundamental mechanisms of ERAD from studies both in yeast and mammalian cells. We will also discuss recent findings that highlight the contributions of ERAD and ubiquitination machinery to the control of ER functions and association with proximal organelles.
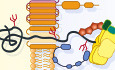
The fundamentals of ERADThe ERAD pathway of both yeast and mammalian cells is comprised of multiple branches, each operating in parallel through a membrane-embedded ubiquitin ligase complex. Each ERAD branch (or complex) engages substrates through recognition domains located in either the lumen or membrane of the ER, facilitating substrate retrotranslocation (or relocalization) into the cytosol where the ubiquitination machinery resides. Soluble secretory proteins confined entirely by the ER lumen must (at least partially) unfold in order to traverse the lipid bilayer for ubiquitination. Both mono- and polytopic membrane substrates, with domains already exposed to the cytoplasm, are afforded the opportunity to be ubiquitinated even before retrotranslocation of their membrane and luminal domains (Fig 1A). Once ubiquitinated by their respective ubiquitin ligase complex, substrates of all ERAD branches converge on a cytosolic AAA-ATPase known as Cdc48 in yeast and p97/VCP in mammals, which aids in the process of retrotranslocation by engaging and “pulling” ubiquitinated substrates from the grasp of the ER membrane, before finally handing them off to the 26S proteasome for degradation.

Figure 1. The steps and ubiquitin ligases involved in ERAD
(A) General steps involved in the ERAD of different classes of misfolded proteins (in black). Ubiquitin ligase complexes in the ER membrane promote the recognition, retrotranslocation, and ubiquitination of ERAD substrates. Recognition of misfolded glycoproteins in the ER lumen requires trimming of glycans (in red, see text for details). Ubiquitinated substrates are released from the ER membrane into the cytosol by the Cdc48/p97/VCP ATPase, trimmed, and re-ubiquitinated before being handed to the proteasome for degradation. (B) E3 ubiquitin ligases and their cognate E2 enzymes characterized in yeast and mammalian cells. Homologous ERAD complexes in yeast and mammalian cells are shown together, separated by dotted lines. ERAD complexes exclusive to yeast and mammalian cells are show in separate boxes. Arrows indicate the subcellular environment by which substrates have been shown to access each E3.
While all ERAD branches are thought to abide by this generic framework, the distinct mechanisms by which most ERAD branches select, differentiate, and retrotranslocate substrates remain largely unknown (see below). The key role of 26S proteasomes as the ultimate destination in substrate destruction in ERAD has long been established (Ward et al, 1995; Hiller et al, 1996). Similarly, the essential role Cdc48/p97/VCP plays upstream to efficiently deliver substrates to the proteasome in ERAD is well documented, with its genetic disruption or pharmacological inhibition causing ERAD substrates to accumulate (Bays et al, 2001; Ye et al, 2001; Jarosch et al, 2002; Rabinovich et al, 2002; Chou et al, 2011). Positioned between the ubiquitin conjugation machinery at the ER membrane and cytosolic proteasomes, Cdc48/p97/VCP provides a common avenue for the continuous processing and transfer of ERAD substrates.
Cdc48/p97/VCP access to substrates emerging from the ER is enhanced by its direct recruitment to ER membranes through factors containing VCP-interacting motifs. In yeast, an N-terminal UBX domain of the ER membrane protein Ubx2 serves as the Cdc48 tether (Neuber et al, 2005; Schuberth & Buchberger, 2005). Likewise, in metazoans, UBX domains as well as other p97/VCP-interacting motifs (e.g., SHP, VIM, VBM) can be found within ERAD complex components such as Derlin-1/2 and UBXD8, and even within E3s themselves (e.g., Hrd1, gp78/AMFR) (Ballar et al, 2006; Morreale et al, 2009; Greenblatt et al, 2011). By recruiting Cdc48/p97/VCP to sites of retrotranslocation, these domains could be seen to facilitate continuous processing of ERAD substrates, expediting their removal from the ER and their delivery to proteasomes. However, not all ER-resident E3 complexes enriched for p97/VCP appear to contain canonical VCP-interacting motifs (Fenech et al, 2020). It is unclear whether yet unidentified interacting motifs are present within these ERAD branches, or whether the polyubiquitin adducts on substrates themselves might be interacting directly with p97/VCP cofactors such as Npl4 (Stein et al, 2014). IP-MS studies have revealed that association of p97/VCP with ER-resident E3s varies greatly, being highly enriched in a subset of E3s while absent or only very modest for a few other E3s (Hülsmann et al, 2018; Fenech et al, 2020). One interpretation could be that p97/VCP enrichment with ER-resident ubiquitin ligases serves as an indicator of substrate flux through individual ERAD branches. In this scenario, ER-resident E3s that are not enriching p97/VCP may be performing functions that are beyond the scope of protein degradation. Deciphering the variety of roles being played here by ER-resident E3s will be an important area for future studies.
Just passing through: ERAD substrate engagement with Cdc48/p97/VCPAlthough the general role of Cdc48/p97/VCP in ERAD has been appreciated for some time, several important recent advances have helped to understand how this ATPase is able to engage diverse substrates originating from all ERAD branches (Fig 1A and B) as well as other cellular pathways.
Cdc48/p97/VCP is an abundant and multifunctional AAA-ATPase. Monomers of Cdc48/p97/VCP assemble to form a hexamer with two stacked rings (D1/D2) defined by a central pore. Cdc48/p97/VCP recruits the co-factors Npl4/Ufd1, which bind asymmetrically to one side of the ring (cis/D1). Npl4/Ufd1 facilitates recruitment of polyubiquitinated substrates to the complex by interacting directly with their ubiquitin chains (Sato et al, 2019). Interestingly, Npl4 interaction reportedly leads to the unfolding of one of the ubiquitin molecules within the chain as it inserts into a Npl4 groove (Cooney et al, 2019; Twomey et al, 2019). This unfolded ubiquitin molecule can then reach the central pore of the Cdc48/p97/VCP ring through which the substrate is threaded (Fig 2A). Translocation of substrates through the Cdc48/p97/VCP central pore is powered by the aromatic residues that line it, which change conformation upon ATP hydrolysis, as has been observed in related ATPases including the 19S proteasome (de la Peña et al, 2018) and bacterial Clp ATPases (Martin et al, 2008; Rizo et al, 2019). Coordination of the ATPase cycles of the six Cdc48/p97 monomers within the ring allows the aromatic residues to function in a “staircase” or “conveyor belt” fashion, resulting in the effective translocation of oligo-ubiquitinated substrates all the way through the pore of a Cdc48/p97/VCP hexamer to the opposite (trans/D2) side of the ring. Since the binding and translocation of the substrate is initiated by its ubiquitin moiety, the Cdc48/p97/VCP complex can process large and diverse groups of substrates, provided they are ubiquitinated as is the case for ERAD substrates (Ji et al, 2021). In parallel, Cdc48/p97/VCP may also serve as a “retrochaperone” for ubiquitinated membrane substrates, effectively promoting their solubility in the aqueous cytoplasm following retrotranslocation (Neal et al, 2017). This feature may be more important in yeast than in metazoans, as the latter also possess the multi-subunit Bag6-SGTA-Trc35 complex, which similarly engages and promotes solubility of retrotranslocated substrates (Wang et al, 2011; Xu et al, 2012; Payapilly & High, 2014).

Figure 2. Proposed mechanisms for key processing steps in ERAD
(A) Cdc48/p97/VCP ATPase (yellow) has a universal role in pulling ubiquitinated substrates from the ER membrane. Ubiquitin molecules conjugated to the substrate interact with the ATPase complex and unfold as they enter the central pore (1). Rounds of ATP hydrolysis promote the threading of the substrate through the central pore (2). As substrates emerge on the trans side of the ATPase complex, they may be re-ubiquitinated by soluble ubiquitin ligases (3). De-ubiquitinating enzymes (Otu1 and Yod1 in yeast and mammals, respectively) on the cis-side remove residual ubiquitin moieties, facilitating the remaining residues of the substrate to travel through the central pore. Glycans conjugated to glycoproteins are removed by peptide N-glycanase enzymes (Png1 and NGly1 in yeast and mammals, respectively) associated with the cis- or trans-sides of the Cdc48/p97/VCP complex. (B) Proposed mechanisms for the retrotranslocation of luminal glycoproteins. In yeast (from left to right), upon recognition by Hrd3/Yos9, substrates engage with the membrane domains of Hrd1 and Der1, which form two halves of a channel. Besides being rich in hydrophilic residues, Hrd1/Der1 membrane regions promote the local thinning of the ER membrane, thereby facilitating the retrotranslocation of the substrate to the cytosolic side of the ER membrane, where it is ubiquitinated by the activity of Hrd1 RING domain (in inset). Ubiquitinated substrate is pulled through the channel and across the membrane by the Cdc48/p97/VCP as described in (A). Although less mechanistic studies are available, analogous steps are thought to occur during the retrotranslocation of luminal glycoproteins in mammals (Right to left). (C) Proposed retrotranslocation mechanism for a membrane ERAD substrate in yeast. Membrane substrates ubiquitinated by the Hrd1 or Doa10 ERAD ubiquitin ligase complexes interact with Dfm1, which facilitates their retrotranslocation with the assistance of the Cdc48/p97/VCP ATPase complex. Dfm1 can also promote retrotranslocation of ubiquitinated substrates independently of Hrd1 and Doa10. The mechanisms and the oligomeric state of Dfm1 during retrotranslocation are unclear.
Along with substrates, the cis-side of the Cdc48/p97/VCP complex also scaffolds other components such as yeast Otu1 and mammalian Yod1. These deubiquitinating enzymes have been shown to shorten the polyubiquitin chains on ERAD substrates, likely easing their translocation through the Cdc48/p97/VCP complex (Ernst et al, 2009; Stein et al, 2014), and may also prevent re-binding of already translocated substrates (Ji et al, 2021). The association of other DUBs, such as Atx3, with Cdc48/p97/VCP suggests that ubiquitin chain trimming is a routine process for this platform (Wang et al, 2006). In mammalian cells, substrates emerging on the trans-side of the Cdc48/p97/VCP complex with shortened ubiquitin chains are re-ubiquitinated by association with the chaperone BAG6 and the ubiquitin ligase RNF126 (Hu et al, 2020). Other cytosolic ubiquitin ligases, such as UBE3C and UBR4, also appear able to act on ERAD substrates, and may serve a similar role (Leto et al, 2019; van de Weijer et al, 2020). They increase the efficiency of degradation of some ERAD substrates by adding branched and mixed ubiquitin chains, which facilitate fast access to the proteasome.
An outstanding conundrum is how N-linked oligosaccharides, covalently modifying most putative ERAD substrates while in the ER lumen, could navigate through Cdc48/p97/VCP. Their extended structures would presumably hinder progress of a polypeptide through the narrow central pore. A solution to this apparent obstacle may come from recruitment of the cytosolic deamidating enzyme peptide N-glycanase (NGly1) to Cdc48/p97/VCP by way of its PUB domain (Allen et al, 2006; Zhao et al, 2007). The NGly1 PUB domain binds to the D1 and/or D2 ATPase domains of p97/VCP, placing it at least in the vicinity of retrotranslocated (and glycosylated) substrates. If present, this enzyme could ensure that N-linked sugars conjugated to most membrane and secreted proteins while in the ER are removed after retrotranslocation, but prior to engagement by Cdc48/p97/VCP. Consistent with this model, inhibition of the proteasome causes retrotranslocated ERAD substrates to accumulate in a deglycosylated form in the cytosol (Wiertz et al, 1996). However, removal of N-linked glycans is not essential, as yeast cells lacking Png1 peptide N-glycanase, or mammalian cells treated with the NGly1 inhibitor zVAD-fmk, are still capable of degrading glycosylated ERAD substrates, albeit at lower efficiency (Suzuki et al, 2000; Grotzke et al, 2013). Whether glycans are in this case threaded through the pore, or slide in between the subunits of the Cdc48/p97/VCP hexamer as recently observed for certain ubiquitin molecules, is unclear (Ji et al, 2021).
Parallel operations: how different ERAD branches divide the loadThe ability for a wide and diverse range of substrates to be targeted by ERAD comes from its organization into multiple branches, each operating in parallel but with distinct specificity. Yet, the molecular determinants for dividing the labor among ERAD branches and the mechanisms of substrate preference and recognition of each branch remain, for the most part, elusive. It was first proposed that in yeast, with a simple ERAD system comprised of only three branches (Fig 1B), the differential selectivity for misfolded proteins is based on the compartment in which the lesion/misfolding occurs (Huyer et al, 2004; Vashist & Ng, 2004; Carvalho et al, 2006). This model posits that substrates exposing misfolded domains to the ER lumen (ERAD-L substrates) or to the cytosol (ERAD-C substrates) engage with ERAD branches using Hrd1 or Doa10, respectively. However, even in this simple eukaryote, the scenario becomes more complex when considering proteins with lesions in the membrane (ERAD-M substrates), with all three yeast ERAD branches (Hrd1, Doa10, and Asi) able to target ERAD-M substrates (Sato et al, 2009; Habeck et al, 2015; Ruggiano et al, 2016; Natarajan et al, 2020).
In mammals, more than 25 different E3 ubiquitin ligases reside in the ER, and at least a dozen of them have been implicated in ERAD (Fig 1B). Despite this expansion of available degradation routes, some aspects of substrate selectivity nevertheless remain evolutionarily conserved. For example, misfolded soluble substrates in the ER lumen (of the ERAD-L variety) are exclusively processed by the HRD1 branch, as in yeast. However, the ERAD-L/M/C classification, based on the location of the misfolded domain, appears an oversimplification when describing the complex and modular nature of ERAD organization in mammalian cells, which may leverage or prioritize other features to influence the engagement with particular ER-E3s (Bernasconi et al, 2010). With membrane-bound substrates, potentially accessible to every ER-E3 through the phospholipid bilayer they share, those intrinsic features seem likely to be important in determining ER-E3 selectivity as well as priority. So, while there will be instances of ER-E3 exclusivity, as in the case of luminal substrates and Hrd1, other substrates are redundantly processed by multiple routes (see the example of HMGCR, below). While the large number of ER-E3s in higher eukaryotes makes deconvolving redundancy challenging, new platforms that multiplex knockdowns/knockouts may help to shed light on functional overlap that may exist. Moreover, the rules and the molecular underpinnings governing the selectivity and preference of each ER-E3 branch for different misfolded membrane proteins presented to them remain undefined. Some clarity into this complex scenario, however, comes from the regulated degradation of certain folded ER-resident membrane proteins by ERAD. As detailed below for certain sterol metabolism enzymes, specific adaptors are involved in recognition and recruitment of substrates to a defined ERAD branch.
Hrd1 ERAD branch: One way out (of the ER lumen)ERAD regulates ER functions primarily via its role in the degradation of membrane proteins. However, the mechanism of ERAD is best understood for luminal (ERAD-L) substrates, which are targeted to the Hrd1 complex in both yeast and mammals. The Hrd1 complex is conserved throughout eukaryotic evolution and constitutes the most extensively studied branch of ERAD. It plays a central role in maintaining ER homeostasis, with many of its key components under the transcriptional control of the unfolded protein response (UPR) (Friedlander et al, 2000; Fenech et al, 2020). Early studies in yeast revealed important aspects on how soluble substrates are recognized in the ER lumen and retrotranslocated across the membrane into the cytosol via a multi-subunit, membrane-spanning complex organized around the Hrd1 ubiquitin ligase (Carvalho et al, 2006; Denic et al, 2006; Gauss et al, 2006). These studies, primarily using the model substrate CPY*, a misfolded version of yeast carboxypeptidase (Hiller et al, 1996), were instrumental in defining the factors and mechanisms used by the Hrd1 complex to select/engage and retrotranslocate misfolded glycoprotein substrates across the ER membrane. As part of a multi-component complex, Hrd1 is associated on either side of the ER membrane with additional proteins that provide functionalities essential for the ERAD process (Fig 2B). The Hrd1-associated factors Hrd3 and Yos9 discriminate and engage misfolded glycoproteins in the ER lumen (Fig 2B). In mammals, this conserved process is carried out by the homologues Sel1L and OS-9/XTP3-B, respectively. Bipartite recognition of adjacent but distinct substrate features by these proteins marks substrate engagement in ERAD: While Hrd3/Sel1L, cooperating with Kar2/BiP, recognize and engage unstructured polypeptide regions, the lectin-like binding domains of Yos9/OS-9 and XTP3-B selectively recognize N-linked glycans with a terminal α1,6-linked mannose residue (Quan et al, 2008; Hosokawa et al, 2009; Xie et al, 2009). In the “mannose timer” model, this glycan modification arises from prolonged exposure to a slow-acting mannosidase enzyme in the ER lumen (Hebert & Molinari, 2012). It has also been proposed that substrate trimming may require traffic through an ill-defined ER-derived compartment containing this mannosidase activity (Benyair et al, 2015). Presence of this trimmed glycan on a nascent polypeptide is indicative of its long ER residence and of unsuccessful cycles of folding/refolding (Hebert & Molinari, 2012; Xu & Ng, 2015). Thus, by incorporating information on the folding state and residence time in the ER, the “degradation signal” recognized by the Hrd1 complex prioritizes terminally misfolded proteins while sparing both folded and newly synthesized molecules from degradation. This substrate recognition mechanism is conserved in mammalian cells, exemplified by studies on the degradation of NHK, the “null Hong Kong” mutant variant of human alpha-1 antitrypsin (Christianson et al, 2008; Hosokawa et al, 2008). However, the presence of trimmed oligosaccharides is not an essential prerequisite, as misfolded, nonglycosylated substrates in the ER lumen are also targeted by the Hrd1 complex. How exactly such substrates are recognized is unclear, but Yos9 (OS-9/XTP3-B) does not appear to play a role (Leto et al, 2019). Instead, work in mammalian cells has shown that the ER luminal chaperones BiP and ERDJ5 as well as HERP, a subunit of the Hrd1 complex and likely the functional homologue of yeast Usa1, appear to be involved (Okuda-Shimizu & Hendershot, 2007).
To reach the ubiquitination machinery, substrates recognized in the lumen must traverse the ER membrane into the cytosol. A path for substrate retrotranslocation across the lipid bilayer was recently revealed by improved cryo-EM reconstructions of the yeast Hrd1 complex (Wu et al, 2020). These structures reveal that the membrane domains of Der1 and Hrd1 each harbor hydrophilic cavities facing the luminal and cytosolic sides of the membrane, respectively, thus forming two “half-channels” (Fig 2B). Lateral gates in the Der1 and Hrd1 halves face each other in a thinned membrane region. Still, Der1 and Hrd1 are not directly docked to each other, suggesting that portions of the substrate may contact bilayer lipids during retrotranslocation. Together with earlier crosslinking experiments (Carvalho et al, 2010; Mehnert et al, 2014), these data support a model in which substrates recognized by Hrd3/Yos9 reach the Der1 luminal cavity in close proximity, subsequently inserting as a loop into the Hrd1/Der1 channel. The membrane thinning induced by Hrd1/Der1 could also be expected to facilitate retrotranslocation by lowering the energy barrier for moving hydrophilic amino acid residues through the hydrophobic interior of a lipid bilayer (Wu et al, 2020). As a portion of the substrate emerges on Hrd1 cytosolic cavity, its ubiquitination by the Hrd1 cytosolic RING domain (and E2s) would promote the engagement of Cdc48/p97/VCP for providing the ratcheting/pulling force necessary for extraction (discussed above). Subsequent rounds of ATPase hydrolysis by this complex then imposes directionality onto later stages of substrate retrotranslocation.
This current model suggests that the active channel is formed by a monomeric Hrd1 complex (Wu et al, 2020). However, biochemical studies on endogenous Hrd1 indicate that it is present in high molecular weight complexes corresponding to dimeric or even higher oligomeric states (Horn et al, 2009; Carvalho et al, 2010; Hwang et al, 2017). Whether these oligomers are involved in processing distinct substrates, or instead correspond to immature, idle, or regulatory forms of the Hrd1 complex, are important open questions. How Hrd1 channel gating is regulated also remains unresolved. Hrd1 channel activity in yeast was shown to be stimulated by self-ubiquitination on one (or more) lysine residues within its RING domain as well as substrate engagement (Baldridge & Rapoport, 2016; Peterson et al, 2019; Vasic et al, 2020). But whether these events cause Hrd1 conformational changes or increase channel “activity” by some other mechanism is currently unknown. It remains to be confirmed whether auto-ubiquitination is also essential for functionality of mammalian Hrd1, which notably has far fewer available lysine acceptors in its cytoplasmic domain, although one lysine within the catalytic RING domain is conserved throughout all metazoans. These efforts to decipher the molecular underpinnings of how Hrd1 complexes process lumenal substrates are slowly bringing the ERAD mechanism into greater focus. New structural techniques and reconstitution systems should help to accelerate this progress, allowing to fully visualize the dynamic nature of ERAD.
Escape Options: Multiple exit routes from the ER membraneAlong with its exclusive role for ERAD-L substrates, the Hrd1 branch of ERAD described above is also actively involved in the degradation of select ERAD-M substrates. Notably however, there are several indications that the degradation path for membrane-embedded substrates through Hrd1 is distinct from that taken by the soluble ERAD-L substrates. First, yeast Hrd1 mutants compromised in their ability to process some ERAD-M substrates remain competent to degrade those of ERAD-L (Sato et al, 2009). Unlike ERAD-L substrates, Hrd1—already co-occupying the same membrane environment—does not necessarily require cofactors in order to engage ERAD-M substrates, and may even play a direct role in their recognition (Sato et al, 2009). Second, retrotranslocation of membrane-bound substrates appears to occur independently of Hrd1 self-ubiquitination (Neal et al, 2018, 2020). Lastly, retrotranslocation of membrane-bound substrates is independent of Der1 (Taxis et al, 2003; Vashist & Ng, 2004). Instead, Dfm1, a rhomboid-pseudoprotease protein related to Der1, and able to replace Der1 in an alternative Hrd1 complex (Goder et al, 2008), has been implicated in the degradation of membrane proteins (Neal et al, 2018). Curiously, Dfm1 also facilitates Hrd1-independent membrane protein retrotranslocation, by assisting other ERAD branches such as the Doa10 branch (Fig 2C) (Stolz et al, 2010; Avci et al, 2014; Neal et al, 2018). In fact, retrotranslocation of an engineered membrane substrate ubiquitinated independently of ERAD ubiquitin ligases appears to depend only on Dfm1 (Neal et al, 2018). A recent structure showed that Derlin1, the mammalian Dfm1 homologue, can assemble into tetramers suggesting that retrotranslocation of membrane proteins by these proteins may involve their oligomerization (Rao et al, 2021). The complex relationship between Hrd1, Der1, and Dfm1 in yeast, and the variations observed, raises intriguing questions of distinct functionalities associated with these factors. This is illustrated by the observation that yeast Dfm1 deletion strains can over time restore membrane protein degradation by duplicating chromosome XV, which contains Hrd1, or simply by overexpressing Hrd1 (Neal et al, 2018). How closely this represents the organization, regulation, and function within mammalian cells is an important question to address in the future, particularly in light of the increased number of participating factors.
The yeast ERAD branch defined by the Doa10 complex, composed of the ubiquitin ligase Doa10 and the ubiquitin conjugation enzymes Ubc6 and Ubc7, was initially identified through its role in degradation of the soluble transcriptional repressor Mat2α (Swanson et al, 2001). Doa10 and its mammalian equivalent TEB4/MARCHF6 also target other soluble proteins, which like Mat2α associate transiently with the cytoplasmic face of the ER via amphipathic helices (Ravid et al, 2006; Stefanovic-Barrett et al, 2018). The Doa10 (or TEB4/MARCHF6) ERAD branch also engages membrane proteins, including various tail-anchored (TA) proteins containing a single transmembrane segment near to the C-terminus (Swanson et al, 2001; Habeck et al, 2015; Stefanovic-Barrett et al, 2018; Dederer et al, 2019). Well-studied examples include unassembled Sbh2, a subunit of the SEC translocon involved in protein import into the ER, and Ubc6, a subunit of the Doa10 complex, implying a level of autoregulation. Interestingly, ERAD degradation of Sbh2 and Ubc6 depends in both cases on their transmembrane region. However, while the tail-anchor region is sufficient for Doa10-mediated degradation of Sbh2, Ubc6 degradation also requires its own ubiquitin-conjugation activity conferred by its cytosolic domain, suggesting that ubiquitin conjugation may occur intramolecularly in this case (Walter et al, 2001). Doa10 further promotes degradation of mislocalized peroxisomal TA proteins such as Pex15 (Dederer et al, 2019; Matsumoto et al, 2019). In this case, Pex15 mislocalized to mitochondria is removed from the outer membrane by the Msp1 ATPase and handed over to Doa10. Whether Doa10 recognizes mislocalized Pex15 while in the cytosol, upon its insertion into the ER membrane, or both is unclear. It is also unclear whether Doa10 recognizes Pex15 through its TA region. While these examples suggest a general role of Doa10 in quality control of TA proteins, our understanding of the molecular details involved in the recognition of these substrates remains incomplete.
Mammalian TEB4/MARCHF6 and a partly redundant ERAD ubiquitin ligase, TRC8/RNF139, are also implicated in quality control of TA proteins. Curiously, recognition by TEB4/MARCHF6 or TRC8 requires the transmembrane (TM) segment of these TA proteins to be cleaved by the intramembrane protease SPP (Signal Peptide Peptidase) (Boname et al, 2014; Chen et al, 2014; Stefanovic-Barrett et al, 2018; Yücel et al, 2019). SPP-mediated processing of TA substrates occurs within the plane of the membrane, likely on the lumen-proximal leaflet, followed by ubiquitination via TEB4/MARCHF6 or TRC8. The large membrane domains of TEB4/MARCHF6 and TRC8, containing 14 and 12 predicted TM segments, respectively, might facilitate the retrotranslocation of the processed TA substrate with the aid of p97/VCP ATPase. It remains unclear how SPP initially selects its TA substrates and how the processed TA proteins are handed off to and subsequently recognized by TEB4/MARCHF6 and TRC8.
In addition to TA proteins, TEB4/MARCHF6 targets other membrane substrates. This includes degradation of the membrane domain of yeast Erg11 (Erg11TM), when expressed in mammalian cells (van de Weijer et al, 2020). In contrast to TA substrates, TEB4/MARCHF6-dependent degradation of Erg11TM may not be redundant with TRC8 nor depend on prior substrate processing by SPP. However, as in the case of TA proteins, the TM segment of Erg11TM was found essential for degradation, suggesting a direct role of TEB4/MARCHF6 in TM domain recognition. TEB4/MARCHF6 is involved in degradation of unassembled FAM8A1, a membrane-bound component of the Hrd1 complex, thus exemplifying co-regulation between ER-E3 complexes (Schulz et al, 2017). How Doa10 and TEB4/MARCHF6 recognize/engage substrates has yet to be established, but it appears to involve a conserved C-terminal element (Zattas et al, 2016). The precise substrate feature(s) being recognized have not been defined either, yet it is apparent that not all TMs are being recognized. For example, CYP51A1TM, a model substrate analogous to Erg11TM and encompassing the TM domain of the human Erg11 homologue, is targeted to ERAD via the recently identified RNF185/MBRL complex and independently of TEB4/MARCHF6 (van de Weijer et al, 2020). At the core of this complex is the multi-spanning membrane protein Membralin (MBRL or TMEM259), which independently binds to the ubiquitin ligase RNF185 and to either TMUB1 or TMUB2, members of the transmembrane and ubiquitin-like domain-containing protein family (van de Weijer et al, 2020). While more work is required to understand how RNF185/MBRL functions, recognition of CYP51A1TM was found to involve both its TM region as well as a short luminal amphipathic helix. This difference underscores how seemingly equivalent substrates have evolved to demand the broad range of metazoan E3s offered by evolutionary diversification.
ERAD and regulation of ER functionsInvestigations of ERAD go beyond the quest for components, their mechanistic organization, or roles in degradation of aberrant proteins. Seminal studies in yeast have shown that levels and stability of the key sterol biosynthesis enzyme, 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR), are controlled by ERAD, leading to the identification of Hrd1 (in fact, HRD stands for HMGCR Degradation) as its central component (Hampton et al, 1996). This early work demonstrated the role of ERAD in sterol homeostasis and its potential to regulate other aspect of ER physiology. Indeed, the search for other endogenous ERAD substrates, their regulation, and physiological relevance, is now an important and growing area of research. In this section, we discuss examples of key membrane proteins targeted by ERAD and how this mode of regulation affects cell physiology.
Regulation of sterol biosynthesis by ERADSterol levels profoundly affect the properties of cellular membranes, and sterol biosynthesis through the mevalonate pathway demands precise regulation (Luo et al, 2020). In addition to cholesterol in mammals and ergosterol in yeast, the mevalonate pathway produces other essential metabolites such as vitamin K, ubiquinone, or steroid hormones. Coordinated synthesis of these various products depends on feedback regulation of rate-limiting mevalonate pathway enzymes by ERAD-dependent turnover (Fig 3). The best-studied cases are mammalian HMGCR and SQLE (squalene monooxygenase), and their respective yeast counterparts Hmg2 and Erg1. In both yeast and mammalian cells, degradation of these enzymes involves two distinct ERAD branches. In yeast, high flux through the mevalonate pathway resulting in accumulation of intermediate metabolites such as farnesyl pyrophosphate, leads to Hmg2 downregulation by Hrd1-dependent ERAD. High levels of geranylgeraniol pyrophosphate appear to destabilize the Hmg2 membrane domain, something that is perceived by the Hrd1 complex as a misfolded membrane protein and thus triggers its rapid degradation (Wangeline & Hampton, 2018). ERAD-mediated regulation of mammalian HMGCR also occurs upon high flux through the mevalonate pathway, but the involvement of distinct signals and components reveals the evolution of more complex controls of this pathway in higher eukaryotes (Schumacher & DeBose-Boyd, 2021). When sterol intermediates abound, HMGCR binds the integral ER membrane protein INSIG, whose association with several ERAD ubiquitin ligases facilitates ubiquitination and subsequent degradation of HMGCR (Sever et al, 2003a, 2003b). The precise identity of the ubiquitin ligases involved in HMGCR degradation has been a point of contention for some time, but recent genome-wide CRISPR/Cas9 knockout screens indicate gp78 and RNF145 as the principal E3s, with only minor contribution by Hrd1 (Menzies et al, 2018). INSIG is itself subject to cholesterol-regulated degradation via the gp78 ERAD branch, revealing an additional layer of complexity of HMGCR regulation (Lee et al, 2006). Furthermore, in the presence of high sterol levels, UBIAD1, a key enzyme in vitamin K biosynthesis, binds to the HMGCR/INSIG complex, attenuating degradation of ubiquitinated HMGCR (Schumacher et al, 2015). Membrane accumulation of the isoprenoid geranylgeranyl pyrophosphate triggers UBIAD1 release from this complex, restoring fast HMGCR degradation. Thus, ERAD-mediated degradation of HMGCR is finely tuned by multiple mevalonate pathway metabolites and regulators.

Figure 3. Regulation of sterol biosynthesis by ERAD
Endoplasmic reticulum-resident enzymes involved in sterol biosynthesis pathways of yeast (top) and mammals (bottom) are shown, along with the different ubiquitin ligases that regulate their abundance via ERAD. Sterol-related biosynthetic products of each enzyme are shown (in blue) as are the nonsterol isoprenoids (in green). Enzymes whose regulation by ERAD has not yet been described are also shown (in red).
Squalene monooxygenase constitutes another point in this pathway subjected to ERAD-mediated feedback regulation. SQLE and Erg1 levels depend upon mammalian TEB4/MARCHF6 or its yeast homologue Doa10, respectively (Foresti et al, 2013; Zelcer et al, 2014). SQLE degradation is stimulated by high cholesterol concentrations and depends on an N-terminal region in SQLE (Gill et al, 2011). Cholesterol-induced membrane association of the SQLE N-terminus in combination with increased stability of the ubiquitin ligase TEB4/MARCHF6 results in accelerated SQLE degradation (Chua et al, 2017; Sharpe et al, 2019). Degradation of yeast Erg1, which does not share the cholesterol-responsive N-terminus of SQLE, is instead stimulated by lanosterol (Foresti et al, 2013). However, the mechanisms sensing lanosterol concentration in the membrane have not yet been identified.
Other sterol-biosynthetic enzymes found to be targeted for ERAD degradation include mammalian DHCR7 (Prabhu et al, 2016), DHCR14 (Capell-Hattam et al, 2020), CYP51A1 (van de Weijer et al, 2020), and yeast Erg3 (Jaenicke et al, 2011), Erg11 (Foresti et al, 2014) and Erg25 (Buck et al, 2020; Christiano et al, 2020). However, the impact of these proteolytic events on sterol homeostasis, or whether additional enzymes in the pathway are also targets for ERAD, remains to be fully ascertained. Regardless, ERAD appears central to the complex regulation of sterol homeostasis necessary for viability in all eukaryotes, which suggests that co-evolution may have occurred to enable the dynamic regulation of this essential biosynthetic pathway.
Border patrol: the maintenance of ER membrane identity
留言 (0)