African violet (S. Jolly Diamond), was used in the present study. All Saintpaulia genotypes were provided from the genotype collection of the Institute GABIT (Genetics and Agricultural Biotechnology Institute of Tabarestan) and the plant material was maintained in greenhouses under controlled conditions. For plant transformation, the leaves were sterilized with 1% sodium hypochlorite for 20 min, washed thoroughly with sterile water, and cultured to germinate on MS basal medium (pH 5.7) containing MS salts [43] 2% sucrose, 0.8% bacto-agar, 1 mg.L−1 6-benzyl amino purine (BA), and 1 mg.L−1 indole-3-butyric acid (IBA). The cultures were then incubated in a growth chamber under the condition of 25±1ºC, a 16-h florescent light (100 µmol/m2/sec), and a 8-h dark cycle. 8 weeks later, young leaves were collected and used for the transformation test.
Transient expression via infiltration of petal
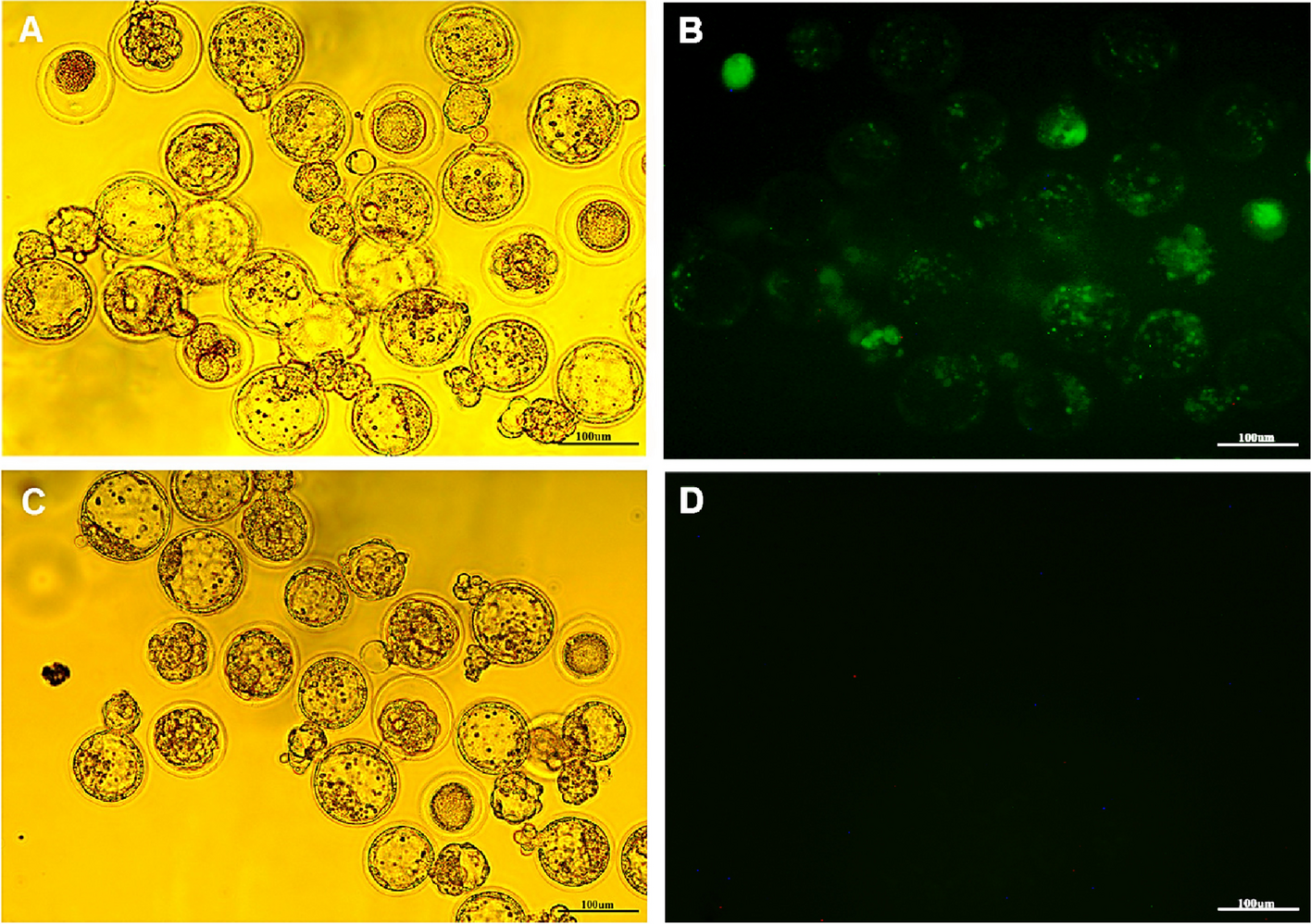
African violet flowers were infiltrated with Agrobacterium tumefactions strain LBA4404 harboring both pBI121 and pCAMBIA1304. For this, the LBA4404 cells were cultured in 10 ml LB broth with antibiotics overnight, pelleted and re-suspended in a medium #1003 (AB media salts + NaH2PO4 240 mg.L−1 + glucose 10 g/l + MES 14.693 g/l) supplemented with 100 µM acetosyringone and cultured for 4 h [35]. The cells were then pelleted and re-suspended to a concentration of A600 = 0.5 in 1% (w/v) glucose solution (PH 5.3) supplemented with 100 µM acetosyringone [35]. Flower buds or opened flowers were pierced with a needle and infiltrated with the Agrobacterium culture using a syringe. Three days after injection and subsequent change in specimen’s conditions, the injected petals (both with and without gene construct) were separated from non-injected ones. After DNA extraction, the existence of two genetic parts was examined using PCR test. To further investigate the color change of petals, the cross section of the injected petals (with and without gene construct) was evaluated using a light microscope.
Stable transformation
A single colony of plasmid-carrying Agrobacterium was picked up and incubated in 10 ml LB medium containing 50 mg.L−1 Kanamycin (Km) and 100 mg.L−1 Rifampin (Rif) at 28˚C overnight. Bacteria cells (OD 600 - 0.5 - 0.6) were then washed and resuspended in AB medium (5 g/l glucose; 1 g/l NH4Cl; 0.3 g/l MgSO4∙7H2O; 0.15 g/l KCl; 10 mg.L−1 CaCl2; 2.5 mg.L−1 FeSO4∙7H2O; 3 g/l K2HPO4; 1.15 g/l NaH2PO4∙H2O) without antibiotics. The cultures were incubated at 28 °C for 6 h to reach mid-log phase, followed by the addition of 100 µl of acetosyringone (AS; SigmaAldrich, St. Louis, MO, USA) and further incubation at 28 °C for 4 h. The bacterial suspension was then centrifuged at 3000 rpm for 10 min, and the pellet was dissolved in MS medium (MS salts; 0.9 mg.L−1 thiamine; 1 mg.L−1 BA; 1 mg.L−1 IBA; 200 mg.L−1 KH2PO4; pH 5.6) supplemented with 100 µM acetosyringone and 5% glucose. The suspension was then diluted to final OD600 0.6-0.8. One day before the infection, leaves were excised from 4- and 6-week-old in vitro grown African violet and incubated on MS solid medium supplemented with 100 µM acetosyringone. Infection was carried out by adding 15 ml of diluted Agrobacterium suspension to the pre-cultured explants for 1 h. Excess Agrobacterium explants were blotted on sterile filter papers to remove excess liquid. The infected explants were transferred on MS solid medium containing 100 µM acetosyringone and 5% glucose, and left to grow for 3 days in a dark condition at 28˚C. Then they were washed 2-3 times with shaking (the first shaking at 220 rpm and subsequent shakings at 100 rpm; 30 min each time) in MS liquid medium with 500 mg.L−1 cefotaxime. Explants were dried with sterile filter papers and transferred to MS medium (MS salts; Nitsch vitamins; 1% sucrose; 1 mg.L−1 BA; 1 mg.L−1 IBA 0.7% bacto-agar; pH 5.8) with 250 mg.L−1 cefotaxime (for inhibition of Agrobacterium growth), 50 mg.L−1 Kanamycin (for selection of pBI121/4’CGT vector) and 75 mg.L−1 Hygromycin (for selection of pCAMBIA1304/AS1 vector). Plates were then incubated at 28˚C in a growth chamber with a 16-h florescent light (100 µmol/m2/sec) and 8-h dark cycle. Explants were regularly subcultured to new MS medium supplemented with 250 mg.L−1 cefotaxime and suitable antibiotic (75 mg.L−1 Hygromycin and 50 mg.L−1 Kanamycin) every two weeks. The single shoot regenerated from inoculated explants was excised from the calli and transferred onto the MS basal medium supplemented with 100 mg.L−1 cefotaxime. Rooted plantlets were then transferred into pots containing 70% peatmoss; 30% perlite and grown in above-mentioned conditions.
CAPS analysisDNA extraction and PCR
Total DNA was extracted from fresh petal of putative transgenic plants with CTAB method as previously described [44]. For PCR analysis, p1 and p2 specific primers were used for 4’CGT and AS1 genes (Supplementary Table S2). PCR was performed using GoTaq® Green Master Mix (Promega, Madison, WI, USA) in a T100 thermal cycle (Bio-Rad), with initial denaturation at 95 ºC for 5 min, followed by 35 cycles at 95 ºC for 45 s, 58 ºC for 30 s and 72 ºC for 2 min, and a final extension step at 72 ºC for 10 min. The analysis of products was performed via 1% agarose gel electrophoresis and sequencing (Sanger Sequencing method, ABI 3730 XL).
Southern blot analysis
For Southern blot analysis, genomic DNA of petals was treated with 10 µg L−1 RNase for 4 h at 25 °C, followed by phenol-chloroform extraction and ethanol precipitation. Representative probes were prepared with digoxigenin, by used DIG DNA labeling and detection kit. 30 µg DNA of each leaves sample was cut with NcoI enzyme for AS1 and BamHI enzyme for 4CGT (Thermo Fisher) by incubating for 16 h at 37 °C. The digested products were separated by 1% agarose gel, denatured in 1.5 M NaCl, 0.5 M NaOH for 30 min each, and transferred to hybridization membrane (GeneScreen, DuPont, Boston, MA, USA). Hybridization was performed at 65 °C with digoxigenin-labeled probe (Amersham Rediprim II, GE Healthcare, Pittsburgh, PA, USA). After 20 h of hybridization, the membrane was washed twice in 2× SSC at room temperature for 15 min each, twice in 2× SSC, 1% SDS at 65 °C for 30 min each, and finally once in 0.1× SSC at room temperature for 30 min. The membrane was exposed to an imaging plate at room temperature and the signal was detected by a phosphor imager (Typhoon FLA 7000, GE Healthcare, Pittsburgh, PA, USA).
Quantitative realtime PCR (qRTPCR) for the expression analysis of 4’CGT and AS1 genes
For real-time quantitative PCR, total RNA was extracted from African violet white (non-transformation), transgenic A. violet, and A. majus petals all treated with DNase I, as previously described by wang et al. [45]. Next, cDNA was synthesized from total RNA (100 ng) using the Superscript III First-Strand Synthesis System (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA) and oligo (dT) 20. The transcript levels of 4’CGT and AS1 were analyzed via RT-qPCR (Applied Biosystems 7900HT Fast Real-Time PCR System) using Power SYBRTM Green PCR Master Mix (Applied Biosystems, Thermo Fisher Scientific) according to the manufacturer’s instructions. Transcript levels were calculated based on ∆∆Cq (formerly ∆∆Ct) method [46] using Actin (accession number: AB596843.1) gene as references. Statistical significance of the differential expression levels was assessed as independent experiments (with mean centering and autoscaling) [47]. The Tukey–Kramer test at the 1% level was used for the analysis of aurone biosynthesis-related genes. The results are presented as the standardized mean of SE.
Light Microscopy analysis
The morphology of regenerates transgenic and non-transgenic petals were examined under a VH-Z75 light microscope. Petals were cut into 5 × 5 mm pieces and embedded in 4% agarose. Thin sections (thickness, 100 mm) were cut with a Micro Slicer DTK-1000 (D.S.K.). Sections were examined using a VH-Z75 light microscope (Keyence).
Aureusidin 6-O glucoside identification by HPLC-DAD-MSn
The African violet white, transgenic African violet, and A. majus petals (12 mm) were evaluated separately. Aurone analyses were performed using an Agilent HPLC series 1200 equipped with ChemStation software, a degasser, quaternary pumps, autosampler with chiller, column oven, and diode array detector. The guard column operated at a temperature of 35 C. The mobile phase consisted of 0.1% TFA⁄water (eluent A) and 90% acetonitrile in 0.1% TFA⁄water (eluent B) at a flow of 0.8 mL⁄min using the following gradient program: 20% B (0–3 min); 20–60% B (3–20 min); 60% B isocratic (20–27 min); 60–90% B washing step (27–30 min); and equilibration for 10 min. The total run time was 40 min. The injection volume for all samples was 10 L. Specific wavelengths were monitored separately at 400 nm for aurone and 360 nm for flavones. Additionally, UV⁄Vis spectra were recorded at 520 nm for anthocyanins. The HPLC system was coupled online to a Bruker (Bremen, Germany) ion trap mass spectrometer fitted with an ESI source. Data acquisition and processing were performed using Bruker software. The mass spectrometer was operated in positive ion mode and auto MSn with a scan range from m⁄z 100 to 1000. HPLC-grade acetonitrile, water, trifluoroacetic acid (TFA), naringenin, and chalcone standards were purchased from Sigma (St. Louis, MO). All standards were prepared as stock solutions at 10 mg⁄mL in methanol and diluted in water except for chalcone, which was prepared in 50% methanol. UV external standard calibration was also used to obtain calibration curves of cyanidin-3-O-glucose, naringenin-7-O-rutinoside, and chalcone, which were used to quantify anthocyanins, flavones, and chalcones, respectively. UV external calibration of maritime was employed for the quantitation of AOG.





留言 (0)