PBMC isolation
Blood samples from healthy male volunteers (age range, 18–21 years) were collected into an EDTA anticoagulant tube. For precision and mass accuracy analyses, blood samples were pooled from 20 individual samples. First, the blood samples were diluted with PBS (1/1 v/v), carefully loaded onto a Ficoll gradient, and centrifuged at 400×g for 30 min at room temperature. PBMCs were collected at the interface and washed three times with PBS (at 400×g for 10 min). Next, monocytes were purified by positive selection using specific monoclonal antibodies, anti-human CD14 magnetic particles (Catalog No. 557769; BD Biosciences, CA, USA) coupled to magnetic beads. Finally, positive cells were resuspended in RPMI-1640 medium (Hyclone, UT, USA) containing penicillin and streptomycin. The study was approved by the Ethics Committee of the Xi’an Jiaotong University Health Science Center (No. 2018–485).
Cell culture
Monocytes isolated from blood were seeded in cell culture plates at a density of 1 × 106 /mL and grown in RPMI 1640 medium containing 10% fetal serum (v/v), penicillin (100 units/mL), and streptomycin (100 μg/mL) and kept at 37 °C in an atmosphere of 5% CO2. To induce macrophages, monocytes were cultured for 7 d in the presence of 100 ng/mL recombinant human macrophage colony-stimulating factor (M-CSF) (Catalog No. 300–25, PeproTech, France). The purity of monocytes and macrophages was evaluated by flow cytometry. For foam cell formation, macrophages were incubated with 50 μg/mL ox-LDL (Yiyuan Bio-technologies, Guangzhou, China) in culture medium for 48 h, and oil red O staining was used to identify whether foam cells were successfully induced.
Flow cytometry
To detect the efficiency of monocyte transformation into macrophages, monocytes and macrophages were incubated with specific antibodies against CD11b (Catalog No. 561688, BD Biosciences), and positive cells were detected using flow cytometry. In brief, cells were harvested, washed, and the cell suspension was adjusted to a concentration of 1 × 106 cells/mL in ice-cold PBS, 10% fetal calf serum, and 1% sodium azide. We added 1 μg/mL of PE-conjugated CD11b to the cell suspension, which was then incubated for 30 min in the dark at room temperature. The cells were washed three times, centrifuged at 400×g for 5 min, and resuspended in ice-cold PBS, 10% fetal calf serum, and 1% sodium azide. Cells were stored in the dark at 4 °C until analysis.
Oil red O staining
As foam cells uptake lipids during their formation, oil red O staining was used to identify foam cells. After the culture medium was discarded, cells were washed twice with PBS and fixed with formalin for 10 min. The solution was rinsed with PBS for 1 min and then with 60% isopropanol for 15 s. Cells were then exposed to oil red O for 1 min in the dark at 37 °C, rinsed with 60% isopropanol for 15 s, and then washed three times with PBS for 3 min each. Finally, after being sealed, cells were observed under a Nikon light microscope.
Protein extraction and digestion
Cell samples were sonicated three times on ice using a high-intensity ultrasonic processor (Scientz) in lysis buffer (8 M urea, 1% Protease Inhibitor Cocktail). The remaining debris was removed by centrifugation at 12,000×g at 4 °C for 10 min. Finally, the supernatant was collected, and the protein concentration was determined using a BCA kit according to the manufacturer’s instructions. For digestion, the protein solution was reduced with 5 mM dithiothreitol for 30 min at 56 °C and alkylated with 11 mM iodoacetamide for 15 min at room temperature in the dark. The protein sample was then diluted by adding 100 mM TEAB to a urea concentration of less than 2 M. Finally, trypsin was added at a 1:50 trypsin-to-protein mass ratio for the first digestion overnight and 1:100 trypsin-to-protein mass ratio for a second 4 h digestion.
TMT labeling and HPLC fractionation
After trypsin digestion, the peptides were desalted using a Strata X C18 SPE column (Phenomenex, CA, USA) and vacuum-dried. The peptide was reconstituted in 0.5 M TEAB and processed according to the manufacturer’s protocol for the TMT kit. The tryptic peptides were fractionated into 18 fractions by high pH reverse-phase HPLC using Agilent 300 Extend C18 column (5 μm particles, 4.6 mm ID, 250 mm length). Briefly, peptides were separated into 80 fractions with a 2 to 60% acetonitrile gradient in 10 mM ammonium bicarbonate at pH 10, over 80 min. Then, peptides were combined into 18 fractions and dried by vacuum centrifuging. Three replicates per condition were performed. Nine TMT labels (126, 127 N,127C,128 N, 128C, 129 N, 130C, 131) were used per run and 18 TMT runs were performed.
LC-MS/MS analysis
The tryptic peptides were dissolved in solvent A (an aqueous solution containing 0.1% formic acid and 2% acetonitrile) and directly loaded onto chromatographic column ReproSil-Pur Basic C18 (1.9 μm particles, 100 μm ID, 25 cm length). The gradient ranged from 9 to 26% solvent B (an aqueous solution containing 0.1% formic acid and 90% acetonitrile) over 40 min, 26 to 35% in 14 min, and was increased to 80% in 3 min, then hold at 80% for the last 3 min, all at a constant flow rate of 350 nL/min on an EASY-nLC 1000 UPLC system.
The peptides were subjected to NSI source (the standard source accompanying the Q Exactive TM Plus) followed by tandem mass spectrometry (MS/MS) in Q Exactive™ Plus (Thermo) coupled online to the UPLC. The electrospray voltage applied was 2.1 kV. The m/z scan range was 350 to 1800 for a full scan, and intact peptides were detected in the Orbitrap at a resolution of 70,000. Peptides were then selected for MS/MS using the NCE setting as 28, and the fragments were detected in the Orbitrap at a resolution of 35,000. The data-dependent procedure that we performed alternated between one MS scan followed by 20 MS/MS scans with a 15.0 s dynamic exclusion. Automatic gain control (AGC) was set at 5E4. The fixed first mass was set to 100 m/z.
Database search
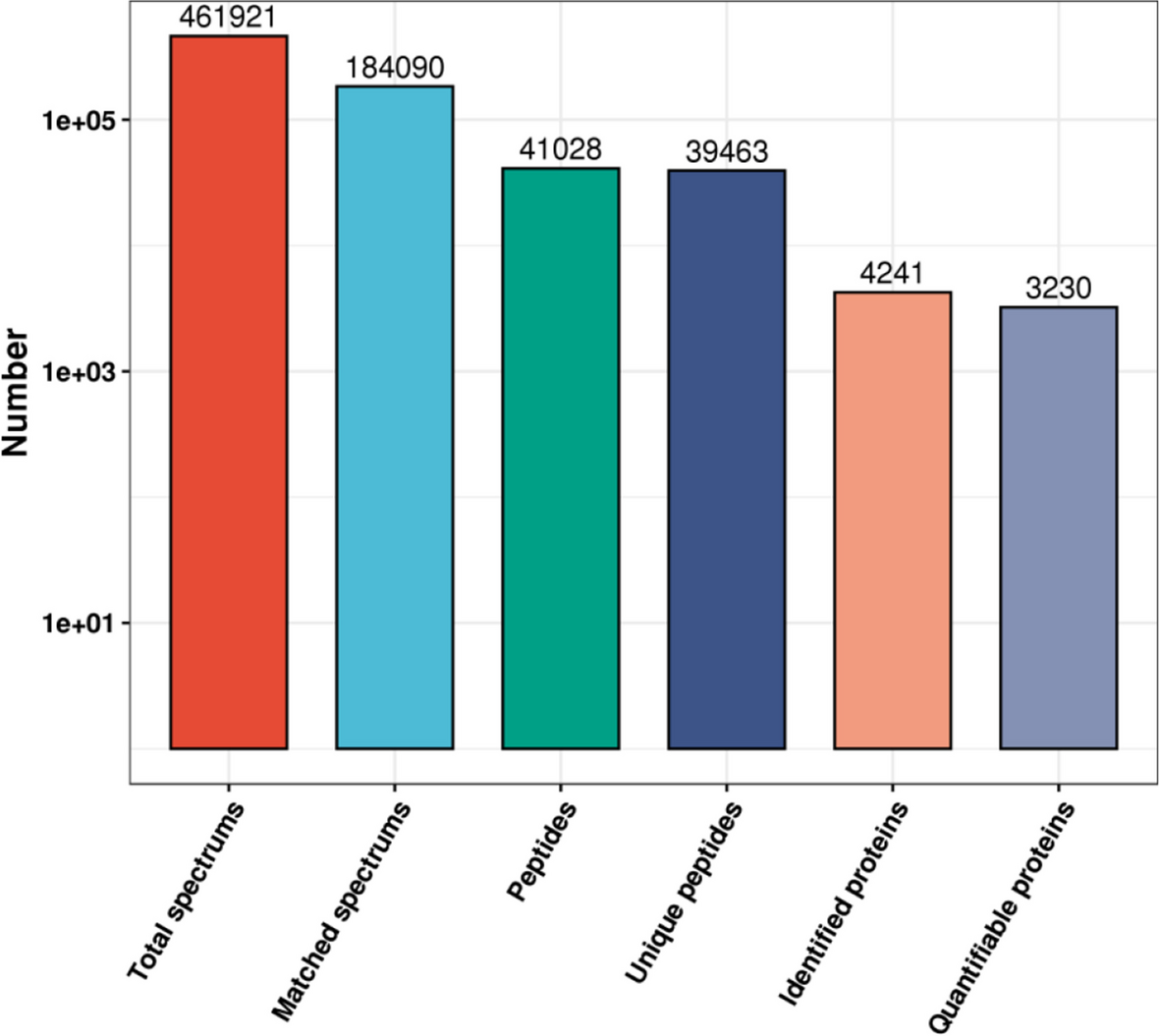
The resulting MS/MS data were processed using the MaxQuant search engine (v.1.5.2.8). Tandem mass spectra were searched against the human SwissProt database (downloaded on 16 August 2018) concatenated with a reverse decoy database. Trypsin/P was used as a cleavage enzyme, allowing up to two missing cleavages. The mass tolerance for precursor ions was set as 20 ppm in the first search and 5 ppm in the main search, and the mass tolerance for fragment ions was set as 0.02 Da. Cys carbamidomethyl was specified as a fixed modification, and Met acetylation and oxidation were specified as variable modifications. FDR was adjusted to < 1%, and the minimum score for modified peptides was set at > 40.
Bioinformatics analysis
The proteomic results were analyzed using multiple approaches. The Gene Ontology (GO) annotation proteome was derived from the UniProt-GOA database (http://www.ebi.ac.uk/GOA/). Wolfpsort, a subcellular localization prediction, was used to predict subcellular localization. The Kyoto encyclopedia of genes and genomes (KEGG) database was used to annotate protein pathways. First, the KEGG online service tool, KAAS, was used to annotate the protein’s KEGG database description. The annotation results were mapped to the KEGG pathway database using the KEGG online service tool, KEGG mapper. GO annotation and KEGG database were used to identify DEP enrichment by a two-tailed Fisher’s exact test to test the enrichment of the DEPs against all identified proteins. Protein-protein interaction (PPI) network analysis was conducted using STRING version 11.0 (https://string-db.org/). The STRING-generated network was visualized and edited using Cytoscape version 3.8.2.








留言 (0)