
記住我
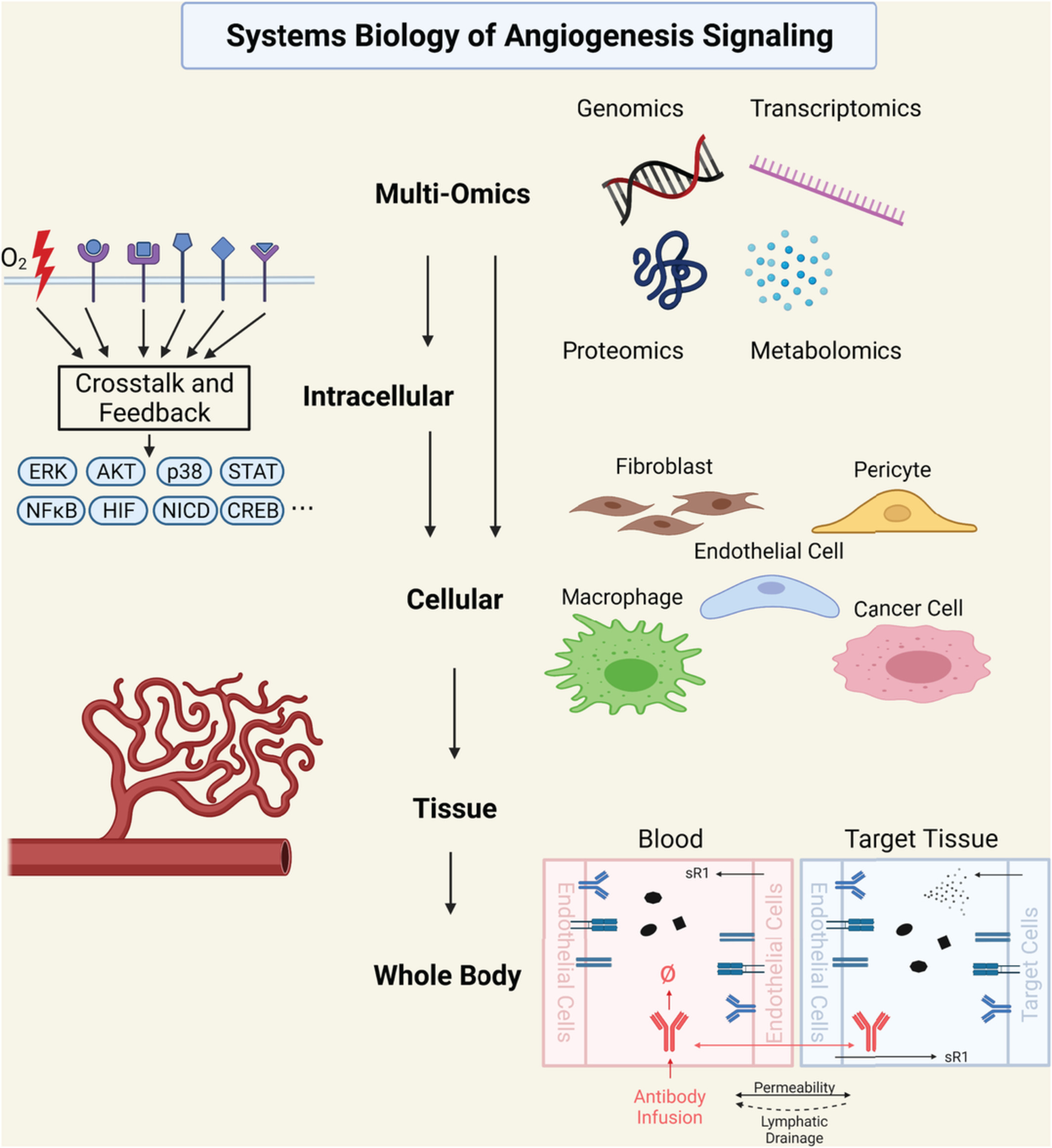

Angiogenesis, the formation of new blood vessels from preexisting vessels, is a quintessential biological process widely involved in development, growth, wound healing and reproduction (Carmeliet & Jain, 2011; Eelen et al., 2020; Flegg et al., 2020; Logsdon et al., 2014). Angiogenesis by nature is a multiscale process and is tightly regulated by many molecular and cellular mechanisms and mediators (De Palma et al., 2017). For example, tissues experiencing lack of oxygenation can initiate hypoxia response via the intracellular hypoxia-inducible factor (HIF) pathway and potently induce angiogenesis (de Heer et al., 2020; Semenza, 2012). Endothelial cells, among the many cellular regulators of angiogenesis, drive angiogenesis through processes including cell proliferation, migration, tip-stalk cell selection, and cellular signaling which controls vascular permeability and stability (W. Chen et al., 2019; Gaengel et al., 2009; Lamalice et al., 2007). In complex tissue microenvironments in human diseases (e.g., cancer, ischemic diseases), a spectrum of biological signals is systematically integrated to determine the degree of angiogenic activities, which ultimately contributes to disease progression and resolution (Beck & Plate, 2009; De Palma et al., 2017; Johnson et al., 2019).
Dysregulation of angiogenesis is a phenomenon widely observed and implicated in the pathophysiology of various major human diseases. Carmeliet identified over 70 angiogenesis-dependent diseases (Carmeliet, 2005) and the list has been growing. In tumors, excessive and abnormal angiogenesis (e.g., disorganized, leaky vessels) is critically involved in tumor growth and metastasis (Carmeliet & Jain, 2011). In the eye, pathological angiogenesis is an important contributing factor to vision impairment in many ocular diseases such as diabetic retinopathy and age-related macular degeneration, affecting millions worldwide (Campochiaro, 2013). In ischemic diseases such as coronary artery disease (CAD), peripheral arterial disease (PAD), myocardial infarction (MI), and cerebral ischemia, insufficient formation of stable and nonleaky blood vessels is a pivotal reason behind the persistent damage and lack of natural regeneration of the ischemic tissue (Dragneva et al., 2013; Iyer & Annex, 2017; Yoo & Kwon, 2013). In infectious diseases, which include COVID19, the role of angiogenesis and endothelial dysfunction has been well recognized; more generally, angiogenesis and its associated molecular factors are key players in the immune response (Osherov & Ben-Ami, 2016; Smadja et al., 2021). In regenerative medicine and tissue engineering, creating potent vascular networks is a crucial element in the optimal application of implants (Rouwkema & Khademhosseini, 2016). Since angiogenesis is innately a multi-pathway, multifactorial, and multiscale process, the systems biology modeling approach, which mechanistically integrates knowledge and data across scales and studies, is particularly useful to help decipher the complex molecular/cellular interactions and emergent pathway/network features that dynamically regulate angiogenesis in human health and disease (G. Liu et al., 2011; Logsdon et al., 2014). Here, we review examples of systems biology modeling efforts toward the quantitative integrative understanding and translational investigation of angiogenesis from the following four aspects: intracellular signal transduction pathways of quintessential angiogenesis modulators (e.g., HIFs, VEGFs, Angiopoietins, FGFs), multi-pathway cell-level players involved in angiogenesis (e.g., endothelial cells, macrophages), blood vessel sprouting and formation, and tissue-/body-level characterization of angiogenesis and its implications in human diseases. We also discuss efforts that utilize multi-omics data-driven approaches to decipher novel angiogenesis drivers and core angiogenesis-related pathway networks. Our review emphasizes the mechanistic relevance and significance of these systems biology models in the multiscale pathophysiology of angiogenesis-related human diseases as well as the translational and therapeutic insights delivered by such modeling efforts. One of the themes is on cancer as cancer progression involves a wide spectrum of complex nonlinear cell signaling and communication. Of note, while the emphasis is on the various signaling pathways, here we will not dive deeply into the important mechanotransduction phenomena that affect angiogenesis, as there are excellent reviews devoted to the subject (Flegg et al., 2020; Murfee et al., 2015).
2 MODELS OF MAJOR CELLULAR/INTRACELLULAR INDUCERS OF ANGIOGENESISIn this section, we discuss mechanistic models built to better understand some key inducing pathways of angiogenesis, including intracellular regulation of HIF and eNOS as well as the signal transduction networks of several growth factor-mediated receptor pathways. While studying the unknown nonlinear emergent properties of known biological networks are usually of primary interest to modeling researchers, it can certainly be envisioned that the systems biology modeling framework and strategy can also be integrated with omics-based high-throughput data to predict and identify new molecular regulations, mechanisms, and hypotheses.
2.1 HIF stabilization and HIF-mediated cellular pathways in angiogenesisHypoxia is a key driver of angiogenesis, and HIFs are the master regulators in the cellular hypoxic response (Krock et al., 2011; Semenza, 2012). At the molecular level, functional HIFs are heterodimers composed of the α and β subunits, while the α subunits (e.g., HIF1α, HIF2α) would typically undergo oxygen-dependent protein degradation in normoxia. The biological processes that regulate HIFα stabilization and degradation under different oxygen tensions include a number of biochemical reactions with various interacting molecular species forming a highly dynamic system, and translational therapeutic modulations of HIF activity by targeting these reactions/species have been extensively investigated as an angiogenesis-based strategies in cancer and ischemic diseases (Majmundar et al., 2010). Below we discuss systems biology computational models specifically formulated to advance the understanding of the different mechanistic aspects of HIF stabilization and HIF-mediated cellular pathways in angiogenesis (selected model information is also summarized in Table 1).
TABLE 1. Summary of selected computational systems biology models of major intracellular signaling pathways in angiogenesis, including HIF stabilization and HIF-mediated cellular pathways, eNOS regulation, and growth factor-mediated pathways Pathway/axis modeled Cell type modeled Relevant disease areas Summary of model objectives/findings References HIF stabilization and HIF-mediated signaling pathways Hypoxia-HIF1α General General Showed that variations in intracellular molecular environment would result in different HIF1α expression patterns (Qutub & Popel, 2006) Hypoxia-HIF1α General/validated experimentally in HEK293 cells General Showed that the different mechanistic functions of PHD and FIH would result in a nonlinear relationship between HIF1α protein stability and its transcriptional activity (Nguyen et al., 2013) HIF-microRNA-VEGF Endothelial cells Cancer, PAD Simulated cellular production of VEGF under hypoxia and microRNA regulation; proposed a mechanistic explanation for insufficient VEGF response in ischemic vascular diseases (Zhao & Popel, 2015) HIF-TGFβ-microRNA-TSP1 Endothelial cells, fibroblast Cancer, PAD Simulated cellular TSP1 production under the control of hypoxia, TGFβ signaling and microRNAs; evaluated in silico strategies that can therapeutically modulate production of TSP1 under relevant pathological conditions (Zhao et al., 2017) HIF1α-ROS General Cancer, ischemia Explained how ROS affects HIF regulation through opposite mechanisms and the resulting differences in the HIF response in tumor and ischemia/reperfusion (Qutub & Popel, 2008) HIF1α-p53 General General Characterized reciprocal mechanistic regulation between HIF1a and p53 under different hypoxic conditions (P. Wang, Guan, et al., 2019) Regulation of eNOS function VEGF/VEGFR2-calcium-eNOS/cGMP Endothelial cells Cancer Simulated targeted interventions to inhibit eNOS activity under high tissue VEGF concentrations in tumor (Q. Wu & Finley, 2020) Insulin-MAPK/endothelin1-PI3K/eNOS Endothelial cells Diabetes Characterized differential changes of eNOS and endothelin1 activities under different pathological conditions (Muniyappa et al., 2020) Shear stress-eNOS Endothelial cells General Characterized multilevel regulation of eNOS function under fluid shear stress and evaluated the impact of different interventional strategies (Koo et al., 2013) eNOS uncoupling Endothelial cells General Investigated a wide spectrum of mechanisms relating to oxidative stress-induced eNOS uncoupling and proposed model-based strategies to restore eNOS function (Joshi et al., 2017)Growth factor-mediated intracellular signaling pathways
VEGF, sVEGFR1 Endothelial cells PAD Used a mechanistic model of sVEGFR1 to quantitatively explore its mechanism of acting as ligand trap and heteromerization with VEGFR (F. T. Wu et al., 2009) VEGF, sVEGFR1 Endothelial cells General Used a PDE-based model of sVEGFR1 secretion and distribution to show its effect in coordinating blood vessel formation stages (Chappell et al., 2016) VEGF isoforms Endothelial cells PAD, general Demonstrated the effect of the splice isoform of VEGF, VEGF165b's effect on VEGFR2 signaling through VEGFR1 interactions (Clegg et al., 2017) VEGF, TSP-1 Endothelial cells General Using a rule-based model to demonstrate the role of TSP-1/CD47 interaction on VEGFR2 signaling and downstream activation of AKT/ERK and intracellular calcium (Bazzazi et al., 2017; Bazzazi, Zhang, Jafarnejad, Isenberg, et al., 2018) VEGFR-Integrin Endothelial cells General Used a rule-based model of integrin-VEGFR2 interaction to investigate potential mechanisms of integrin-targeted therapies (Bazzazi, Zhang, Jafarnejad, & Popel, 2018) Ang-Tie, sTie2 Endothelial cells General Simulated the effect of soluble Tie2 acting as a ligand trap of Ang1 and compared its effects with engineered ligand trap using an ODE-based model (Alawo et al., 2017) Ang-Tie Endothelial cells General Used a mechanistically detailed models of Ang/Tie signaling pathway, its molecular mechanisms and junctional localization to identify potential mechanistic targets for Tie2-targeted therapy (Y. Zhang et al., 2019; Y. Zhang, Kontos, et al., 2021) FGF2 General General Proposed a kinetic model of FGF and its complex formation with FGFR and HSPG (Ibrahimi et al., 2004) FGF-FGFR Endothelial cells General Used a PDE-based model to demonstrate the effects of FGF triad formation and its intracellular responses (Filion & Popel, 2004) FGF-2 Myocardium Cardiovascular disease Investigated the distribution and retention of FGF-2 following exogenous FGF administration using a compartmental model (Filion & Popel, 2005) FGF-2 General General Used a finite element model of FGF diffusion and reaction to simulate FGF binding kinetics under fluid flow (Patel et al., 2013) HGF Hepatocytes Cancer Used a combination of quantitative and qualitative modeling to identify and validate a signaling network of HGF-stimulated Akt and ERK activation (D'Alessandro et al., 2015) HGF Cancer cells, cancer-associated fibroblasts Cancer Developed a PDE-based model of the HGF/c-Met signaling between cancer-associated macrophages and tumor cells in the tumor microenvironment (Konstorum & Lowengrub, 2018) HGF Hepatocytes Cancer Tested the synergism of combination therapies targeting the HGF signaling axis with mechanistic model of the HGF/cMet signaling pathway (Jafarnejad et al., 2019) 2.1.1 Intracellular regulation of HIF1α by prolyl and asparaginyl hydroxylasesPhysiological interaction between HIFα subunits and intracellular hydroxylases (e.g., PHDs, FIH) is a primary regulatory step for the subsequent recognition and targeted proteasomal degradation of HIFα by VHL-containing ubiquitin ligase complexes. A series of computational models have been proposed and analyzed to study HIF stabilization (Nguyen et al., 2013). Among those, the work by Qutub et al. is the first mechanistic model that included kinetic details of PHD2 binding with its cofactors such as iron, ascorbate, 2-oxoglutarate, and oxygen (Qutub & Popel, 2006). The model concluded that the hypoxia-induced HIF1α response dynamics could vary significantly depending on different molecular compositions of those cofactors within the cell, which suggested that interventions targeting HIF1α hydroxylation to modulate HIF1α activity may also have different effectiveness across human cell lines. Later, the model developed by Nguyen et al. further incorporated details of FIH-mediated HIF1α asparagine hydroxylation and negative feedback through HIF1-mediated PHD production, together to describe the transcriptional activity of HIF1α under various physiological and pharmacological perturbations (Nguyen et al., 2013). Using their model, the authors predicted an unexpected biological feature regarding the diverging regulation of HIF1α stabilization and HIF1α transcriptional activity when both PHD and FIH are inhibited; further, this seemingly counterintuitive prediction was validated by their experimental results and was explained mechanistically by an emergent model-based regulatory axis.
2.1.2 Complex interactions between HIF1α and p53The tumor suppressor protein p53 is a well-known transcription factor that has wide impacts on cell cycle, apoptosis, senescence, DNA repair and metabolism (Joerger & Fersht, 2016). It is also stabilized under severe hypoxia and can participate in HIF regulation and be regulated by HIFs via different routes (Obacz et al., 2013). On this topic, two related computational models were developed to mechanistically describe the multimodal regulatory cross talk and feedback between HIF1α and p53 pathways under hypoxia (P. Wang, Guan, et al., 2019; Zhou et al., 2015). In the latter model, by performing model stability analyses, the authors proposed a potential bifurcation between predominant intracellular activation of p53 versus HIF1α based on different severities of hypoxia which might serve as an essential axis that regulates the dynamic and coordinated determination of cell fate and pro-angiogenic activation during hypoxia.
2.1.3 HIF1α-ROS interactionsReactive oxygen species (ROS), a class of oxygen-based cellular metabolic products, can also participate in the regulation of HIFs. Experimental studies have shown that PHDs, FIH, and HIFs can be regulated by ROS through a number of direct and indirect mechanisms (reviewed in Acker et al., 2006; Wong et al., 2013). Using the systems biology approach, Qutub and Popel developed a computational model based on mass-action kinetics and incorporated four possible mechanisms of HIF1α-ROS interaction to explain some seemingly contradictory findings in literature on how ROS regulates HIF1α (Qutub & Popel, 2008). The authors analyzed the model simulations under various concentration combinations of five different molecular species that have functional ties to ROS-HIF1α (O2, iron, ascorbate, succinate, 2-oxoglutarate) and provided a mechanistic basis to better understand the differential HIF1α time-course response influenced by ROS in tumors versus in non-tumor ischemia.
2.1.4 HIF-mediated cellular production of pro- and antiangiogenic factorsHypoxia, through the activation of HIFs and other transcription factors, has been shown to regulate the cellular synthesis and secretion of a number of pro- and anti-angiogenic molecules (e.g., VEGF, Angiopoietins, MMPs, TSP1) to control angiogenesis (Krock et al., 2011). Our group has developed two computational models that investigated the dynamic cellular synthesis of VEGF and TSP1, respectively, under the coordinated control of hypoxia, cytokine signaling, and posttranscriptional regulation (e.g., by microRNAs; Zhao et al., 2017; Zhao & Popel, 2015). Driven by thorough quantitative calibration against time-course experimental data as well as focused analyses using clinical and pathophysiological data, the models were then used to propose novel mechanistic explanations for the disrupted angiogenic balance in specific human diseases (e.g., suppressed TSP1 expression in tumors due to oncogenic activation of Myc, insufficient induction of VEGF in PAD calf muscle due to dysregulation of microRNA let-7). Moreover, the models have been used to provide quantitative model-based evaluation of various pathway-targeted therapeutic interventions under disease-relevant pathological conditions in vitro.
2.2 Regulation of eNOS function and its impact in the modulation of angiogenesisEndothelial nitric oxide synthase (eNOS or NOS3) is the predominant producer of nitric oxide (NO), an important vasodilator and regulator of angiogenesis, in endothelial cells. Activation of eNOS is regulated by multiple growth factor-driven pathways (e.g., VEGF, HGF, Insulin), calcium-dependent pathways, as well as fluid shear stress (Fukumura et al., 2001; Koo et al., 2013; Makondo et al., 2003; Muniyappa et al., 2020). Below we discuss systems biology models that were developed to characterize the complex signaling network of stimulation-induced eNOS functional activation and explore its potential therapeutic implications.
2.2.1 eNOS activation regulated by VEGF/VEGFR2 and calciumUsing rule-based modeling, Bazzazi et al. formulated the first mechanistic model that describes eNOS phosphorylation by the PI3K/AKT and calcium/calmodulin axes downstream of VEGF/VEGFR2 pathway (Bazzazi, Zhang, Jafarnejad, Isenberg, et al., 2018). Using the model, the authors tested the influence of the anti-angiogenic matricellular protein thrombspondin-1 (TSP1), which has been implicated in the pathophysiology of PAD and can associate with cellular CD47 to disrupt VEGF/VEGFR2 signaling, on VEGF-induced eNOS phosphorylation. They demonstrated that eNOS activation, compared to AKT, is particularly sensitive to the inhibitory effect introduced by TSP1 and may require targeting of both CD47 and TSP1 to rescue its normal activation in angiogenesis (Bazzazi, Zhang, Jafarnejad, Isenberg, et al., 2018). Later, the work by Wu and Finley further advanced this rule-based framework and incorporated the chaperone protein HSP90 as well as multiple activation states of eNOS and its downstream signal transduction involving arginine, NO, sGC (soluble guanylate cyclase) and cGMP (cyclic guanosine monophosphate; Q. Wu & Finley, 2020). They then analyzed the model under different VEGF concentrations that resembled actual VEGF levels present in the tumor microenvironment and evaluated the therapeutic potential of several new intracellular targets/processes in terms of shutting down VEGF-induced pro-angiogenic activation of eNOS and cGMP.
2.2.2 eNOS activation regulated by shear stressAn integrated systems biology model has been developed by Koo et al. to investigate the multilevel regulation of eNOS induced by fluid shear stress from four different aspects: calcium influx and eNOS activation, AKT activation and eNOS phosphorylation, eNOS production, and functional eNOS complex formation (Koo et al., 2013). This mechanistic platform-style model was able to describe time-course experimental data of shear stress-induced eNOS/NO production on the scales of minutes to hours. The model also enabled direct quantitative comparison of functional eNOS generated from two different routes (calcium versus phosphorylation) during shear stress stimulation and their relative contribution to NO production in endothelial cells. Furthermore, the authors also implemented model-based interventions that target two different transcription factors as well as two different modalities that inhibit AKT and characterized their respective influences on shear stress-induced eNOS activation. This is an example of model-based quantitative characterization of mechanical stress-induced endothelial cell signaling; more thorough discussion of multiscale modeling efforts applied toward a better understanding of the mechanical aspects of angiogenesis can be found in Flegg et al. (2020) and Murfee et al. (2015).
2.2.3 eNOS uncouplingIn endothelial cells, oxidative stress can cause eNOS dysfunction (eNOS uncoupling) by depleting tetrahydrobiopterin (BH4), an essential cofactor for eNOS. This will shift eNOS from a NO-producing enzyme to a superoxide-producing enzyme, which would further potentiate the preexisting cellular oxidative stress and endothelial dysfunction (Luczak et al., 2020). To quantitively and dynamically describe the set of complex biochemical reactions governing the process of eNOS uncoupling, Kavdia's group, through a series of systems biology studies, has developed a comprehensive computational model that mechanistically incorporated molecular-level interactions between eNOS, BH4, oxidized biopterins, SOD (superoxide dismutase), L-arginine, oxygen, NO, CO2, ROS, and reactive nitrogen species (Joshi et al., 2017; Kar et al., 2012; Kar & Kavdia, 2011). Using this model, the authors unveiled that as oxidative stress increases beyond a threshold level, eNOS can transition from the coupled state into an uncoupled state accompanied with oscillations of decreased NO production over time. Model analyses also suggested that BH4 supplementation combined with strategies to reduce oxidative stress would be an optimal therapeutic approach to improve eNOS coupling and endothelial dysfunction in diseases (Joshi et al., 2017).
2.3 Growth factor-mediated signaling pathways that drive angiogenesisGrowth factors are secreted protein molecules that bind and activate receptors to regulate physiological processes including angiogenesis. Growth factor-mediated intracellular signaling pathways control various aspects of angiogenesis, including vascular permeability, maturation, quiescence, and stability (Potente et al., 2011). Growth factors and their receptors are regulated by a variety of molecular mechanisms, including ligand secretion and expression, receptor multimerization, ligand competition, receptor trafficking and turnover, co-receptor interactions, downstream regulation of receptor expression, and many other pathway specific regulatory mechanisms (Lemmon & Schlessinger, 2010). The growth factor/receptor interactions, their regulatory mechanisms, and downstream signaling form complex reaction networks that warrant the use of integrative computational systems biology models to gain quantitative understanding of the behavior of the signaling pathway. Below we discuss selected computational models of growth factor-mediated signaling pathways (summarized in Table 1). It should be noted that some of the studies of FGF and HGF pathways described below were conducted in application to non-endothelial cells, for example, fibroblasts and hepatocytes; however, the models are applicable to endothelial cells, with appropriate calibration.
2.3.1 VEGFThe vascular endothelial growth factor (VEGF) signaling pathway is a major signaling pathway regulating vascular growth, maintenance, and remodeling. The VEGF signaling pathway also mediates calcium signaling patterns in endothelial cells associated with proliferation and migration (Noren et al., 2016). The VEGF ligand-receptor system consists of ligands VEGF-A, VEGF-B, VEGF-C, VEGF-D, and placental growth factor (PlGF), receptors VEGR1, VEGFR2, and VEGFR3, and co-receptors neuropilins NRP-1 and NRP-2 (Mac Gabhann & Popel, 2008). In turn, the alternative slicing of VEGF-A gives rise to at least 16 different isoforms with distinct signaling properties, as reviewed in Peach et al. (2018). VEGF signaling pathway is also regulated by TSP-1 and its interaction with CD47 (Kaur et al., 2014), expression of the soluble VEGF receptor 1 (sVEGFR1/sFLT1; F. T. Wu et al., 2010a), splicing isoforms and extracellular regulation of VEGF (Vempati et al., 2014), as well as integrin binding (Somanath et al., 2009). The complexity of the signaling pathway and its myriad of regulatory mechanisms have given rise to computational models that focus on different aspects of the pathway, reviewed in Finley et al. (2015) and Logsdon et al. (2014). F. T. Wu et al. (2009) and Chappell et al. (2016) used ODE-based and PDE-based models, respectively, to quantitatively explore the effect of sVEGFR1 in pathological states and in different stages of blood vessel formation. Wu et al. used a compartmental model of VEGF, sVEGFR1, and their interaction with VEGFR2 to investigate the mechanisms of sVEGFR1's inhibition of VEGF signaling, including it acting as a ligand trap and its heterodimerization with VEGFR (F. T. Wu et al., 2009). Chappell et al.'s model incorporated both inhibitory mechanisms of sVEGFR1 in an integrated PDE-based model of VEGF and VEGFR interaction that demonstrates the stage-specific effects of VEGF during different stages of vascular morphogenesis (Chappell et al., 2016). Clegg et al.'s model focused on the splice isoform VEGF165b and its effect on VEGFR2 signaling through competition of binding to VEGFR1 (Clegg et al., 2017). Mac Gabhann et al. used an ODE-based model, combined with experimental validation, to demonstrate that heterodimers of VEGFR1 and VEGFR2 form at the expense of homodimers at the cell surface, resulting in distinct downstream signaling transduction activities (Mac Gabhann & Popel, 2007). In a more integrative model, Bazzazi et al. used rule-based modeling to construct the complex reaction networks formed by VEGF, its receptors and co-receptor, the interaction with TSP1/CD47, and downstream signaling through AKT, ERK, and intracellular calcium to demonstrate the potential effects of targeting the TSP1/CD47 axis, and the potential effectiveness of combination of pro-angiogenic therapies to rescue VEGF signaling to Akt and eNOS involving this strategy (Bazzazi et al., 2017; Bazzazi, Zhang, Jafarnejad, Isenberg, et al., 2018). In another model, Bazzazi et al. also investigated the potential molecular mechanisms of the interaction of αVβ3 integrin and VEGFR2 and predicted the effects of integrin-targeting therapeutics in inhibiting angiogenesis (Bazzazi, Zhang, Jafarnejad, & Popel, 2018).
2.3.2 Ang/TieThe Angiopoietin (Ang)/Tie signaling pathway is a major endothelial signaling pathway regulating vascular permeability, stability, and quiescence (Eklund & Saharinen, 2013). The Ang/Tie ligand receptor system consists of angiopoietins Ang1-4, receptor Tie2, and co-receptor Tie1 (Leppanen et al., 2017), and is regulated by receptor multimerization (Kim et al., 2005), phosphatase VE-PTP (Winderlich et al., 2009), receptor trafficking and junctional localization (Saharinen et al., 2008), and receptor extracellular domain cleavage (Singh et al., 2012). Computational systems biology modeling allows the complex integration of reaction network formed by the interactions of Ang1 and Ang2 with Tie2, and the molecular regulatory mechanisms. The extracellular domain of Tie2 is known to be cleaved off from the surface of the endothelial cells in inflammatory conditions, forming soluble form of the receptor that can bind and inhibit Ang ligands (Findley et al., 2007). Alawo et al. developed an ODE-based computational model to simulate the effect of soluble Tie2 and an engineered ligand trap on the inhibition of Tie2's activation (Alawo et al., 2017). Our group has also developed ODE-based models, using rule-based modeling, of the Ang/Tie signaling pathway that included detailed molecular mechanisms such as receptor multimerization and clustering, ligand competition and co-receptor binding, receptor trafficking, turnover, extracellular domain cleavage and degradation, junctional localization and downstream signal transduction, and regulation by the VE-PTP (Y. Zhang et al., 2019; Y. Zhang, Kontos, et al., 2021). The models identified that the expression of VE-PTP, the presence of Tie1, and its junctional localization are key molecular events modulating the context-dependent agonistic function of Ang2 and provided a mechanistically detailed mechanism to resolve the controversial roles of Tie1 on Tie2's activation. The models also identified inhibiting the extracellular domain cleavage of Tie2, inhibiting VE-PTP, and promoting Tie1's junctional localization as potential therapeutic strategies to promote vascular stability.
2.3.3 FGFThe fibroblast growth factor (FGF) signaling pathway, consisting of FGF-2 and receptors FGFR1-4, is another important signaling pathway regulating angiogenesis, wound healing, and tissue repair (Bikfalvi et al., 1997). The FGF signaling pathway is regulated by the release pattern of FGF, heparan sulfate proteoglycans (HSPGs), and complex formation between FGF, FGFR, and HSPGs (Bikfalvi et al., 1997). Ibrahimi et al. proposed an ODE-based, kinetic model of the FGF-2/FGFR1/HSPG complex assembly based on surface plasmon resonance data (Ibrahimi et al., 2004). Filion et al. used a PDE-based reaction–diffusion model of FGF-2 and its interaction with cell surface receptors to demonstrate the effect of FGF-2 dimerization and the assembly of triads (FGF-2/FGFR1/HSPG) and double triads (2 FGF-2/FGFR1/HSPG) on the intracellular signaling response of the FGF signaling pathway (Filion & Popel, 2004). Another model by Filion et al. used a compartmental model to investigate the effect of distribution and retention of exogenous FGF-2 on its bioavailability (Filion & Popel, 2005). Patel et al. used a finite-element based model to simulate the different dynamics of FGF-2/FGFR/HSPG triad and FGF-2/HSPG or FGF-2/FGFR complex formation following continuous and bolus delivery of FGF-2 under varying flow conditions, and examined the effects of binding stoichiometry, binding site density, fluid flow, delivery dose, and delivery mode to inform experimental studies on FGF-2 delivery (Patel et al., 2013). In addition to the role of FGF signaling in angiogenesis, other computational models have also been developed to study the biphasic dose–response of FGF-2 due to the its complex formation dynamics with FGFR and HSPG (Kanodia et al., 2014), as well as its potency in activating ERK signaling in fibroblasts compared to platelet-derived growth factor (PDGF; Cirit & Haugh, 2012).
2.3.4 HGFAdditional signaling pathways, including the hepatocyte growth factor (HGF) signaling pathway, has been demonstrated to play an important role in angiogenesis. Its downstream signaling has been demonstrated to be able to stimulate endothelial proliferation, migration, and vascular morphogenesis (Shojaei et al., 2010). HGF signaling pathway consists of HGF and its receptor, c-Met, and often acts synergistically with the VEGF signaling pathway (You & McDonald, 2008). D'Alessandro et al. used a hybrid approach utilizing both quantitative experimental data and qualitative molecular interaction graph to systematically identify and validate the best model structure of HGF-stimulated signaling of Akt and ERK activation, predicting that the efficient inhibition of the downstream signaling of HGF can be achieved by targeting the combinations of PDK1, PI3k, Met, and ERK (D'Alessandro et al., 2015). Konstorum and Lowengrub used a PDE-based, multispecies model of the tumor microenvironment to simulate the interaction of HGF-secreting cancer-associated fibroblasts with c-Met receptors on tumor cells, predicting that disrupting the HGF/c-Met signaling with anti-HGF or anti-c-Met therapy reduces tumor invasiveness and growth (Konstorum & Lowengrub, 2018). Jafarnejad et al. used an ODE-based, mechanistic computational model of HGF, c-Met, its interaction with integrin, and downstream signaling to simulate the effects of monotherapies and combination therapies targeting the HGF signaling axis (Jafarnejad et al., 2019). The model incorporated omics data to predict patient-specific synergism and antagonism of different combination therapies, identified the potential synergistic efficacy of the simultaneously targeting integrin and HGF, Met, or Raf in hepatocellular carcinoma, and provided a framework to identify patients who could benefit from drug combinations with mRNA expression data (Jafarnejad et al., 2019). It should be noted that the models discussed above focus on hepatocytes and cancer cells but can be adapted to other cell types such as endothelial cells with proper calibration.
留言 (0)