
記住我


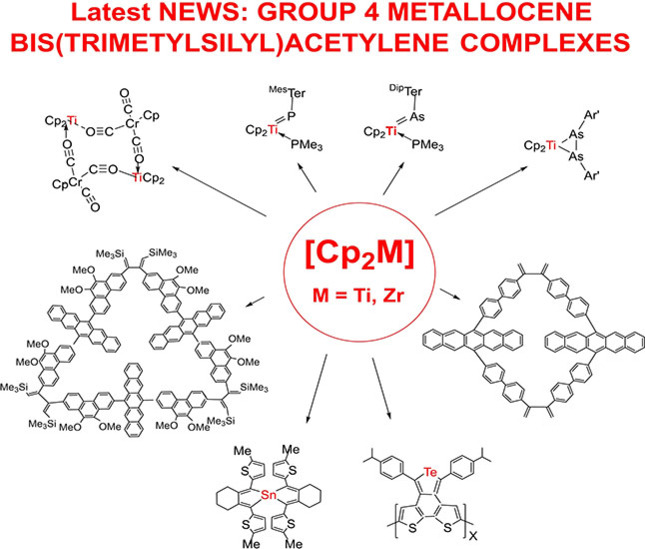
In a recent example from a series of reviews,1 the question whether reactions of complexes such as Cp′2Ti(η2-Me3SiC2SiMe3) and Cp2Zr(py)(η2-Me3SiC2SiMe3) (Cp′=Cp as cyclopentadienyl and Cp*=pentamethylcyclopentadienyl, py=pyridine, Me3SiC2SiMe3=btmsa, bis(trimethylsilyl)acetylene) could be considered as a part of a “Never ending story?” was raised.1f This certainly seems true, as several examples have been added to this story in the relatively short timeframe of the last two years. Some older examples complete the summary, serving as a basis for an understanding of more recent ones. In addition, applications and studies of products obtained from this chemistry are mentioned. Applications, in this context, does not refer to industrial processes, but to the use of these complexes for synthetic purposes. New results for the attractive chemistry of [Cp2TiII] and [Cp2ZrII] have been described in detail. Only parts of the relevant previous work in addition to the current development are briefly mentioned at the beginning of each chapter to show the basis for these extensions. Three distinct parts: (i) substitution reactions of Me3SiC2SiMe3 and coordination of other molecules: (ii) reactions of the new coordination compounds; and (iii) investigations and applications of the coordination compounds for chemical and physical purposes, constitute the main content of the recently published work summarized here. Often, there is overlap between these three areas.
2 Characterization of the Starting ComplexesIn an older study, the question “Are Metallocene-Acetylene (M=Ti, Zr, Hf) Complexes Aromatic Metallacyclopropenes?” was discussed by Jemmis, Jiao and coworkers.2 Therein, the bonding situation for titanocene bis(trimethylsilyl)acetylene complexes Cp2M(η2-Me3SiC2SiMe3) was investigated by computing the titanocene bis-silylacetylene complex Cp2M(η2-H3SiC2SiH3) for M=Ti on the basis of B3LYP density functional theory. The interactions of metals Zr and Hf with alkynes in such complexes have also been calculated. The results of the calculated structural parameters were in excellent agreement with available X-ray molecular structural data. The conclusion of this was the description that the alkyne complexes prefer the metallacyclopropene resonance structure with two in-plane M−C σ-bonds and one out-of-plane π-bond interacting with the metal center. This gives a delocalized three center and two-electron system. After the bonding analysis and the computed stabilization interaction, these alkyne complexes were characterized as aromatic on the basis of the computed nucleus-independent chemical shifts (NICS) at the center of the 3-membered rings over the center of the 3-membered rings.
Very recently, further calculations of such compounds were presented in a paper by Aysin, Leites and Bukalov for a better general understanding of the bonding in 1-heterocyclopropenes regarding the “Aromaticity of 1 Heterocyclopropenes Containing an Atom of Group 14 or 4”.3 In addition to the earlier published results of calculations from the authors for the group 4 metallocene alkyne complexes, structural, electronic (FBO), optical (vibrational spectroscopy), energetic (ISE), and magnetic (NICS scan, GIMIC) criteria for aromaticity were discussed. The obtained results characterize the hetero-cyclopropenes of Si and Ge with aromatic properties as σ* or pseudo-π, whereas all-C-cyclopropenes are σ-aromatic. The bond indices and the aromaticity descriptors ISE as well as the NICS scan show that the silacyclopropene is more aromatic than the germa-cyclopropene. In contrast to these results, the group 4 metalla-cyclopropenes Cp2M(η2-RC2R) with M=Ti, Zr and Hf are not aromatic according to the NICS scan, the GIMIC results and the analysis of the MO bonding scheme. This constitutes a fundamental difference between group 14 and group 4 hetero-cyclopropenes.
3 Substitution of Me3SiC2SiMe3 and Coordination of SubstratesThe substitution of Me3SiC2SiMe3 for substrates like RE=ER to give complexes of the type Cp2Ti(η2-RE2R) and Cp2Zr(L)(η2-RE2R) of main group elements like N, P and Sb has already been described. In a series of investigations, starting from Cp2Ti(CO)24a or Cp2ZrCl2 and Li2[PhN2Ph],4b the complexes Cp2M(L)(η2-PhN2Ph) (M=Ti without L and M=Zr) were obtained, too. Reactions of (rac-ebthi)M(η2-Me3SiC2SiMe3) (M=Ti, Zr, ebthi=ethylene-1,2-bis(5-4,5,6,7-tetrahydro-1-indenyl), Cp*2Ti(η2-Me3SiC2SiMe3), Cp2Ti(η2-Me3SiC2SiMe3) and Cp2Zr(py)(η2-Me3SiC2SiMe3) with PhN=NPh gave, after dissociation of Me3SiC2SiMe3, different complexes of this type, sometimes together with other products.4c
The selective conversion of aryl-substituted triphosphiranes (Ar’3P)3 to titanocene diphosphene complexes Cp2Ti(η2-Ar'P2Ar’2) was observed by Hering-Junghans and coworkers in the reaction with Cp2Ti(η2-Me3SiC2SiMe3) (Scheme 1). This was described for Ar’=Tipp=2,4,6-iPr3C6H2; Dipp=2,6-iPr3C6H2; Mes=2,4,6-Me3C6H2).4d

Formation of titanocene diphosphene complexes Cp2Ti(η2-Ar'P2Ar’).
Some time ago, Breunig and coworkers obtained the complex Cp2Ti(η2-2,6-Mes2C6H3-Sb2-2,6-Mes2C6H3) by the reaction of Cp2Ti(η2-Me3SiC2SiMe3) with 2,6-Mes2C6H3SbH2.4e Very recently, Hering-Junghans and coworkers. extended the series of such compounds and disclosed the analogous arsenic complexes Cp2Ti(η2-Ar'As2Ar’2) with Ar’=Tipp, Dipp, Ter (Scheme 2; Ter=terphenyl).5a

Reactions of Cp2Ti(η2-Me3SiC2SiMe3) with (Ar’−As)3 and Ar'As=AsAr’ to complexes Cp2Ti(η2-Ar'As2Ar’).
In the reaction of Cp2Ti(η2-Me3SiC2SiMe3) with (Ar’-As)3, the complexes Cp2Ti(η2-Ar'As2Ar’) with Ar’=Tipp, Dipp were formed (Scheme 2). The compound Ar'As=AsAr’ with Ar’=Ter reacts with Cp2Ti(η2-Me3SiC2SiMe3) to form such a complex, too. In all these typical reactions, btmsa only acts as a spectator ligand, and by its dissociation, the highly coordinatively and electronically unsaturated reactive 14-electron [Cp2Ti] fragment is formed.5a
In a recently published excellent paper from Hering-Junghans, Reiß and coworkers, the reactions of Cp2Ti(η2-Me3SiC2SiMe3) with phospha- and arsa-Wittig reagents to different products were described (Scheme 3).5b

Reaction products of Cp2Ti(η2-Me3SiC2SiMe3) with phospha- and arsa-Wittig reagents like phosphaindane and PMe3-stabilized phosphinidine as well as arsinidene complexes.
In the reaction with Mes*PPMe3 (Mes*=2,4,6-tri-tert-butylphenyl), the known 3,3-dimethyl-5,7-di-tert-butylphosphaindane was obtained. In the reaction with MesTerPPMe3 (MesTer=2,6-Mes2C6H3), Mes=2,4.6-Me3C6H2) the PMe3-stabilized titanocene phosphinidene complex Cp2(PMe3)Ti=PTerMes was formed (DipTer=2,6-Dip2C6H3). The reaction of Cp2Ti(η2-Me3SiC2SiMe3) with the potential arsa-Wittig reagent DipTerAsPMe3 gave the analogous PMe3-stabilized titanocene arsinidene complex Cp2(PMe3)Ti=AsTerDip. The latter was detected together with diarsene (DipTerAs)2 if substoichiometric amounts of the starting titanium complex were used. The syntheses of the titanocene phosphinidene and arsinidene complexes with Ti=P and Ti=As double bonds was realized, which are best described as singlet biradicaloids (Scheme 3).
Beweries, Reiß and coworkers reported on “Mechanistic Insights into Dehydrocoupling of Amine Boranes using Dinuclear Zirconocene Complexes”.6 In these studies, they were interested in modelling some single reaction steps for the dehydrocoupling and used the complex Cp2Zr(py)(η2-Me3SiC2SiMe3) which, after dissociation of the alkyne and pyridine, serves as a generator for [Cp2Zr]. Reacting Cp2Zr(-CH2SiMe3)(-C≡CSiMe3) with Cp2Zr(η4-butadiene), they obtained a dinuclear hydride-bridged zirconocene complex (Scheme 4). This reaction was not possible when using the alkyne complex Cp2Zr(py)(η2-Me3SiC2SiMe3) instead.

Unsuccessful attempt of using Cp2Zr(py)(η2-Me3SiC2SiMe3) to obtain the dinuclear hydride-bridged zirconocene complex from Cp2Zr(−CH2SiMe3)(−C≡CSiMe3).
If the structurally similar Cp2Zr(−Me)(−C≡CSiMe3) was employed instead of Cp2Zr(−CH2SiMe3)(−C≡CSiMe3), the reaction with Cp2Zr(py)(η2-Me3SiC2SiMe3) allowed the preparation of the corresponding dinuclear hydride-bridged zirconocene complex (Scheme 5).

Successful use of Cp2Zr(py)(η2-Me3SiC2SiMe3) to obtain the dinuclear hydride-bridged zirconocene complex from Cp2Zr(−Me)(−C≡CSiMe3).
4 Substitution of Me3SiC2SiMe3 by Substrates, Coupling Reactions and Investigations of the Obtained New ProductsIn the past, Staubitz and coworkers published a series of highly interesting papers in which the zirconocene complex Cp2Zr(py)(η2-Me3SiC2SiMe3) was used to synthesize tin-containing conjugated heterocycles.7 By intermolecular coupling reactions, several bis(thiophenyl)-substituted octadiynes were converted into zirconacyclopentadienes. During this very effective reaction, functional substituents like iodide stayed intact and the obtained zirconacyclopentadienes reacted by transmetallation to the respective stannole compounds. An interesting example was the reaction of thiophene-substituted stannoles which could be converted to polymers by Stille cross-coupling reactions.7a Using the same method, several other stannoles were obtained.7b During these investigations, it was shown, that the complex Cp2Zr(py)(η2-btmsa) reacted faster and gave higher yields compared to the Negishi reagent. Additionally, functional groups were tolerated by this method, which was applied for several disubstituted alkynes and octadiynes to synthesize the corresponding zirconacyclopentadienes by internal coupling. Under transmetalation conditions with Ar’2SnCl2 (Ar’=−C6F5, −C6H4-p-OMe, −C6H5), several stannoles were obtained.7c, 7d
In several new papers from the Staubitz group, the series of these very interesting investigations was extended to further syntheses.8 Experimental and theoretical studies of a spirostannole and the formation of a pentaorganostannate were reported (Scheme 6).8a The new spirostannole 1,1’,3,3’-tetrakis(5-methylthiophen-2-yl)-4,4’,5,5’,6,6’,7,7’-octahydro-2,2’-spirobi[benzo-c]stannole] was synthesized, following the same procedure of coupling reaction of diynes with Cp2Zr(py)(η2-Me3SiC2SiMe3) and subsequent transmetallation. The results of the geometry optimization by DFT calculations confirm the high planarity, leading to efficient conjugation within the molecule.

Formation of a spirostannole by using Cp2Zr(py)(η2-Me3SiC2SiMe3) and subsequent transmetallation and reaction to a pentaorganostannate.
The obtained spirostannole was characterized as a strongly absorbing material, but an extremely weak emitter in solution. The emission only becomes visible when the solution is cooled. The molecular structure and the electronic behavior of the thermally instable lithium pentaorganostannate was supported by DFT and TD-DFT calculations.
In another recent paper from the same group, the synthesis of four well-defined conjugated polymers, containing stannole units as unusual heterocyclic units in the main chain, was reported.8b The stannole-thiophenyl copolymers were produced by tin-selective Stille coupling reactions in nearly quantitative yields. NMR spectroscopic investigations suggested unaffected tin atoms in the rings. The optoelectronic properties of the iodothiophenyl-stannole monomers and the resulting bisthiophenyl-stannole copolymers were investigated. The molecular structures of several stannoles were studied by single crystal X-ray analysis. Additionally, the influence of the replacement of thiophenes by stannoles in terthiophene and sexithiophene on their optoelectronic and electrochemical properties was investigated.8c Generally, polystannoles with thienyl comonomers are similar to polythiophenes. By using well-defined oligothiophenes as a model to understand the optical and electronical properties of polythiophenes, the team tackled the question of the precise influence of thiophene units in a conjugated backbone of a polymer. These insights were followingly transferred to stannole-containing copolymers. Despite several differences, these materials exhibited a similar behavior to oligothiophenes. Recently, Staubitz and coworkers published a review concerning different methods for the synthesis of stannoles, in which such ring-fused and heteroatom-containing compounds were summarized.8d
Rivard and coworkers had previously synthesized phosphorescent π-extended heteroarenes.9 The reaction of 5,6-didehydro-11,12-dihydrodibenz[a,e]cyclooctyne with Cp2Zr(py)(η2-Me3SiC2SiMe3) yielded zirconacyclopentadienes, which, with (bipy)TeCl2, were converted to “substituent-‘locked’ tellurophenes”. More recently, Rivard and coworkers published a paper in which the complex Cp2Zr(py)(η2-Me3SiC2SiMe3) was used to synthesize tellura(benzo)bithiophenes (Scheme 7).10

Reaction of 5,6-didehydro-11,12-dihydrodibenz[a,e]cyclooctyne with Cp2Zr(py)(η2-Me3SiC2SiMe3) to zirconacyclopentadienes and subsequent conversion with (bipy)TeCl2 to tellurophenes.
Several planar π-extended tellura(benzo)bithiophenes were synthesized with a tellurophene ring fused to a benzobisthiophene. In the 2- and 5-positions of the tellurophenes, aromatic substituents like −C6H4iPr or −C6H4OCH3 are located. The reaction of cumenyl (−C6H4iPr)-substituted tellura(benzo)-bis(thiophene) with p-chloranil led, by oxidation, to Te−C bond cleavage and the formation of an ene-dione (Scheme 8, top). An intramolecular annulation reaction to fuse the tellurophene rings to the aryl groups was only successfully calculated, but not experimentally realized (Scheme 8, center). The bromine-substituted compound reacted with iPrMgCl and [Ni(dppe)(o-tolyl)Cl], via the metalated tellural(benzo)bisthiophene, to a polymer. The data of the obtained products indicated electronic and structural features for phosphorescence in this compound class.

Oxidative cleavage of Te−C bonds to yield an ene-dione; attempted intramolecular annulation; and preparation of a polymer.
In a series of very interesting papers, Tilley and coworkers had reported the use of Cp2Zr(py)(η2-Me3SiC2SiMe3) and Cp2Ti(η2-Me3SiC2SiMe3) for site-selective [2+2+n] cycloadditions.11 The rapid, scalable access to PAHs (polycyclic aromatic hydrocarbons) with alkynyl groups was reported in detail for many examples. In one of these papers, the [2+2+1] addition reaction by Cp2Zr(py)(η2-Me3SiC2SiMe3) with a subsequent transmetalation for the synthesis of alkynylated selenophene-annulated PAHs was described.11e
In a recent paper by Tilley et al., a special zirconocene-mediated cyclic coupling reaction of compounds with two alkynyl groups was published (Schemes 9 and 10).12a In some cases, the zirconocene-catalyzed macrocyclization is reversible through a dynamic C−C bond formation, which was investigated and calculated in detail.12 Additionally, the different obtained products were investigated regarding their X-ray molecular structures and photophysical properties.

Selective dimerization by Cp2Zr(py)(η2-Me3SiC2SiMe3) yielding a macrocycle.

Selective trimerization to a macrocycle by using Cp2Zr(py)(η2-Me3SiC2SiMe3)
The formation of the macrocycles was observed either as a nonselective or as a selective di- or trimerization to yield two geometrically distinct pentacene-containing macrocycles.
For a general understanding of these reactions, Tilley and Miller discussed ligand effects on such zirconacyclopentadiene formations and their reversibility.12b Zirconacyclopentadienes are important intermediates for the synthesis of (E,E)-butadienes, substituted benzenes, thiophenes and conjugated polymers as well as for macrocycles. For this reason, cycloreversion reactions are important for the selective syntheses of these complexes. The authors presented a systematic study of the influence of Cp substituents on the zirconocene coupling reaction of alkynes. Zirconocenes with two cyclopentadienyl (Cp/Cp) and a combination of pentamethylcyclopentadiene/nonsubstituted cyclopentadiene (Cp/Cp*) as well as the ansa-bridged [Me2C(C5H4)2] units were compared. The reversible alkyne coupling of zirconacyclopentadienes with SiMe3 substituents was studied, and the rates of decoupling under the influence of PMe3 were measured. The use of different Cp ligands at zirconium has a significant effect on the rates of the reversibility of alkyne coupling reactions. This is important to get useful information to understand and to optimize the selective coupling to macrocycles and oligomers.
Very recently, Tilley and co-workers reported a new synthetic strategy for the synthesis of new carbon nanobelts (CNBs). Starting from highly fused monomers and using a site-selective [2+2+2] cycloaddition with Cp2(py)Zr(η2-Me3SiC2SiMe3), a high-yielding macrocyclization was realized on large scale.12c The authors concluded that this synthetic method constitutes a general principle for greater synthetic efficiency. The highly fused monomeric building blocks have significant advantages, reducing problems associated with the introduction of strain into the fused-ring macrocycles. The method is described as an important step in the formation of technologically valuable building blocks for more complex materials.
It might be worth mentioning that the authors concluded that the robust synthetic strategies for the synthesis of these two unique pentacene-containing macrocycles were only possible by using the zirconocene coupling strategy.
Norton and coworkers published reactions of the titanium bis(trimethylsilyl)acetylene complexes Cp2Ti(η2-Me3SiC2SiMe3) and Cp*2Ti(η2-Me3SiC2SiMe3) with CpCr(CO)3H (Scheme 11).13a As mentioned before, in these complexes the alkyne dissociates and thus the reactivities of titanocene complexes [Cp2Ti] and [Cp*2Ti] were studied. With CpCr(CO)3H, an addition to form TiIV-H intermediates followed by loss of H2 under formation of bimetallic TiIII-Cr complexes was described.

Mechanism for the reaction of Cp2Ti(η2-Me3SiC2SiMe3) with CpCr(CO)3H.
The formed complexes show a dimeric geometry with a 12-membered Ti2Cr2 ring with bridging CO ligands and Ti−O−C−Cr units. The terminal carbonyls are trans-coordinated to each other. According to DFT calculations, the processes for loss of H2 from the TiIV-H intermediates and the formation of the TiIII-Cr dimer are exergonic. It is somewhat surprising that there was no coupling of CO, as had been observed, for example, by Berry and Bercaw for a bimetallic Zr−Fe complex13b or as it was discussed for other reactions of CO.13c, 13d The same reaction type was described for Cp*2Ti(η2-Me3SiC2SiMe3) and CpCr(CO)3H (Scheme 12).

Reaction of Cp*2Ti(η2-Me3SiC2SiMe3) with CpCr(CO)3H.
In comparison to the above-described reactions of the complexes Cp’2Ti(η2-Me3SiC2SiMe3) with Cp’=Cp, Cp* and CpCr(CO)3H, complex Cp*(C5Me4CH2)Ti(−CH3) reacts with CpCr(CO)3H to yield a TiIV−CH3 species with a coordinated CpCr(CO)3 anion.
5 Reactions of the Formed New Coordination CompoundsUpon coordination of solvents like CH3CN, the observed solvatochromic shifts of the two tetranuclear Ti2Cr2 compounds indicate the breakdown of the dimeric structures (Scheme 13).13a

Coordination of CH3CN to the tetranuclear Ti2Cr2-ring compounds.
The above-mentioned 12-membered heterobimetallic TiIII-Cr complexes showed activity as catalysts for the hydrogenation of an epoxide to yield an anti-Markovnikov alcohol (Scheme 14).13a This was possible without the use of Cp2TiX2 (X=Cl, mesylate), NaCpCr(CO)3, and HCpCr(CO)3 as described in previous catalytic systems from the authors.

Catalytic hydrogenation of an epoxide to an anti-Markovnikov alcohol using the Ti2Cr2 ring compound as catalyst.
6 Recently Published Reviews with Remarks for Reactions of Group 4 Bis(trimethylsilyl)acetylene Metallocene ComplexesIn several recently published reviews, group 4 bis(trimethylsilyl)acetylene metallocene complexes were mentioned or discussed in detail in the context of their special reactivity.
Tonks and coworkers summarized published examples for multicomponent syntheses of 5- and 6-membered aromatic heterocycles by group 4–8 transition metal catalysts.14a They mentioned tetrasubstituted pyrazines as the products of nitriles, magnesium and Cp2TiCl2. For these reactions, 2,5-diazatitanycyclopentadienes were assumed as intermediates which were formed from Cp2Ti(η2-Me3SiC2SiMe3) after alkyne dissociation and tail-to-tail coupling of two nitrile molecules at Cp2Ti.14b, 14c These metallacycles then formed tetrasubstituted several pyrazines through transmetallation from Ti to Al.14d For pyrazine formation, a ring expansion by two further nitrile insertions into the diazatitanycyclopentadienes to 1,3,6,8-tetraazatitana-cyclononatetraenes was assumed. With EtAlCl2,these compounds would, after transmetalation, yield the Et-substituted 1,3,6,8-tetraazaalumina-cyclononatetraenes together with Cp2TiCl2. After hydrolysis, tetrasubstituted pyrazines were obtained in good yields.
In another review, Tamm and coworkers described and compared results of the syntheses and coordination chemistry of heteroatom-substituted alkynes for the example of diaminoacetylenes.14e They mentioned previous results of reactions of dipiperidinoacetylene with titanocene and zirconocene Me3SiC2SiMe3 complexes.14f With metallocenes [Cp2M] (Cp=η5-C5H5), metallacyclopentadienes were formed, whereas the sterically more demanding decamethylmetallocenes [Cp*2M] (Cp*=η5-C5Me5) gave metallacyclopropenes. Group 4 complexes of the heteroatom-substituted alkynes, such as diaminoacetylenes R2NC2NR2, were compared to Me3SiC2SiMe3 complexes. Some of these complexes were synthesized by reduction of Cp’2MCl2 (Cp’=η5-Cp and η5-Cp*) with magnesium in the presence of the diaminoacetylenes. According to NMR spectroscopic studies in solution, the zirconium complexes exist in an equilibrium with the tucked-in tetramethylpentafulvene-diaminovinyl isomer. The latter is formed by an intramolecular CH bond activation and hydrogen transfer.
In a very recent review, Tonks reported details of the progress in Ti-catalyzed and -mediated oxidative amination reactions which formally proceed through TiII/TiIV catalytic cycles.14g He mentioned an older paper about the preparation and regioselective reactions of the zirconocene alkyne complex without stabilizing phosphane ligands Cp2Zr(THF)(η2-tBuC2SiMe3).14h He further summarized and discussed the regiocontrol of the coupling reactions of Me3Si-substituted alkynes, RC2SiMe3, with the partner alkyne. This is strongly influenced by the different alkyne substituents, because the Ti−CSiMe3 bond is stronger than the Ti−CR bond. This was analyzed for other insertion reactions of group 4 metallocenes and used to control regioselectivity in reductive coupling reactions. The above-mentioned regioselective reactions of Cp2Zr(THF)(η2-tBuC2SiMe3) are not directly connected to the here considered titanocene and zirconocene bis(trimethylsilyl)acetylene complexes, but provide insight how, by simple modification of the alkyne substituents, one can modify the coupling selectivity. In principle, this is important for the mentioned coupling reactions of Staubitz7, 8 and Tilley.11, 12 These considerations are also useful regarding the selective syntheses of zirconacyclopentadiene intermediates, which are important for the subsequent clean synthesis of (E,E)-butadienes, transmetallation products, macrocycles and others.14g
Very recently, Fortier and Gomez-Torres described the redox chemistry of low-valent titanium complexes and low-valent titanium synthons.14i They mentioned, among other examples, the previously published reactions of Cp’2Ti(η2-Me3SiC2SiMe3) (Cp’=Cp and Cp*) with water, methanol, phenol, and thiophenol, yielding (Cp2Ti)2(μ-O), Cp*2Ti(OH)2, (Cp2Ti)2(μ-OMe)2, Cp*2Ti(H)(OMe), Cp’2Ti(OC6H4Me)2, and Cp’2Ti(SPh)2, respectively. In addition to this protonation chemistry of Cp’2Ti(η2-Me3SiC2SiMe3), other examples for the formation of divalent titanocene sources [Cp’2Ti] as LVT (low-valent titanium) synthons were summarized. The reactions of Cp’2Ti(η2-Me3SiC2SiMe3) with I2 forms trivalent (Cp’2Ti)2(μ-I)2 and tetravalent complexes Cp’2TiI2.
Cp’2Ti(Me)I species were formed by the oxidative addition of the Me−I bond. The 2,2’-bipyridine radical monoanion Cp*2Ti(bipy), was obtained by the reduction of 2,2’-bipyridine by Cp*2Ti(η2-Me3SiC2iMe3). With 4,5-diazofluorene, 0.5 equiv. of H2 are formed together with an N,N’-bound diazafluorenyl complex. Beckhaus and coworkers extended the chemistry of the Cp’2Ti(η2-Me3SiC2SiMe3) complexes to multinuclear complexes,15 like [(Cp2Ti)3-HATN(Ph)6] (HATN(Ph)6=hexaphenyl-5,6,11,12,17,18-hexaazatrinaphthylene) with multi-electron transfers in the extended π-systems (see below).16 Titanium complexes with multiple titanium−ligand bonds were obtained by the reaction of m-terphenyl azide with Cp2Ti(η2-Me3SiC2SiMe3) to yield terminal imido complexes.
7 Investigations of Former Obtained ProductsAs mentioned above, Beckhaus and coworkers obtained the trinuclear hexaphenyl-5,6,11,12,17,18-hexaazatrinaphthylene-tristitanocene (Cp2Ti)3(μ3-HATNPh6) either through dehydrogenative coupling of 6,7-diphenylquinoxaline in the presence of Cp2Ti(η2-btmsa) or through direct coordination of this fragment to the preformed HATNPh6 ligand (Scheme 15).15

Different formation of (Cp2Ti)3(μ3-HATNPh6) from Cp2Ti(η2-btmsa).
In recent paper by Klüner, Beckhaus, Wittstock and coworkers, the electronic transitions in different redox states of 5,6,11,12,17,18-hexaazatrinaphthylene-bridged complexes were investigated regarding their spectroelectrochemical properties and their quantum chemistry.16 Such multinuclear transition metal complexes, bridged by ligands with extended π-electronic systems, show a variety of complex electronic transitions. While photochemistry and electrochemistry for binuclear complexes was described on several occasions, the related trinuclear complexes are less studied. Hexaphenyl-5,6,11,12,17,18-hexaazatrinaphthylene-tris-titanocene (Cp2Ti)3HATN(Ph)6 shows six oxidation and three reduction waves in the voltammogram. The spectra in solution and the electrochemically formed oxidation products showed electronic transitions in the UV, visible and the NIR ranges. The results of the DFT and linear response time-dependent DFT calculations point at three formally titanium(II) centers with a transfer of an electron to the HATN ligand in the ground state. The optically excited transitions occur only between ligand-centered orbitals. The charged titanium centers provide an electrostatic frame to the extended π-electronic system. The complete active self-consistent field (CASSCF) calculation on a simplified model compound considered the multi-reference character of the three titanium centers. The interpretation of the experimentally observed temperature-dependent magnetic behavior of the different redox states of this compound corresponds to self-assembly reactions of multinuclear complexes mediated by group 4 metallocenes.
Some time ago, coupling reactions of Cp2Ti(η2-Me3SiC2SiMe3) and Cp*2Ti(η2-Me3SiC2SiMe3) with carbon dioxide were investigated.17 With Cp2Ti(η2-Me3SiC2SiMe3), a dinuclear vinylcarboxylate complex was formed which, upon reaction with oxygen, gave a titanafuranone (Scheme 16).

Reactions of Cp2Ti(η2-Me3SiC2SiMe3) with carbon dioxide.
Reactivity and electronic structure analysis of such bimetallic bis-titanocene vinylcarboxylate complexes was published in a very recent paper by Powers, Tonks and coworkers (Scheme 17).18 They reported three new reactions of the dinuclear TiII/TiIII complex [(Cp2Ti)2], characterizing a different reactivity pattern of the reduced dititanium centers. The electronic structure of the obtained compounds was studied by X-ray diffraction and EPR analyses. These electronic structures were used for the covalent bond classification and the electron counting method. Starting from [(Cp2Ti)2], the coordination of CNXyl (Xyl=2,6-(Me)2C6H3) resulted in formation of another bimetallic complex, which after XRD, EPR, and IR analyses was described as a TiII/TiIII electronic structure. The products of reactions with electrophiles were found to depend on the character of the used electrophiles. The reaction with Me3SiCl leads to a monometallic TiIII complex with a chelating α,β-unsaturated silyl ester ligand. The complex was formed by formal substitution of [Cp2Ti] by the trimethylsilyl group. With methyl iodide, a radical reaction was observed, in which oxidation of one titanium to yield a mixed valent TiIII/TiIV complex was observed.

Reactions of the dinuclear vinylcarboxylate complex [(Cp2Ti)2].
These results are in agreement with suggestions that in the bimetallic core a cooperative reactivity is possible through electronic communication. The authors mentioned that their studies about “the fundamental oxidation chemistry of these complexes will hopefully motivate future work on catalytic reactions related to organometallic CO2 coupling using Ti.”
留言 (0)