
記住我


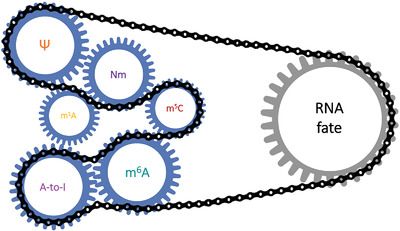
RNA modifications, the so-called epitranscriptome, have emerged as an important regulatory layer of gene expression. Thus far, more than 170 distinct RNA modifications have been identified, distributed among the three kingdoms of life and on all classes of RNA.[1] RNA modifications can control every aspect of RNA metabolism and their dysregulations have been associated with a wide range of physiological alterations and numerous diseases, including neurological diseases, metabolic disorders, and cancer.[2-5] The dynamic nature of some modifications is important to control gene expression upon developmental and environmental changes.[6-8]
tRNAs are the most modified RNA species with up to 25% of all nucleotides carrying a chemical adduct. They are also the RNA species with the largest variety of modifications, in contrast to rRNAs that are decorated mainly by two modification types, pseudouridine and 2′-O-methylation (Nm),[9-12] and only a few additional modifications on the nucleotide base. Modifications on tRNA are important to stabilize its structure, as well to faithfully convey the genetic information carried by the mRNA.[13] In particular, the nature of the modifications present at the anticodon loop can influence the recognition of the mRNA codon and thereby the identity and abundance of the final product. Like tRNA, modifications on rRNA also serve to stabilize RNA-RNA as well as RNA-proteins interactions. These modifications are enriched at the active sites that catalyze peptide bond formation and peptide release, highlighting their importance.[12]
In addition to abundant non-coding RNA, a couple of modifications were also found on mRNA. The most abundant are N6-methyladenosine (m6A) and inosine (I), whereas others such as 5-methylcytidine (m5C), pseudouridine, N1-methyladenosine (m1A) and ribose methylations (Nm) are less represented.[14] Their precise abundance is still under debate as the tools used to quantitate and locate them on transcripts are imperfect. For instance, it is virtually impossible to purify a pure population of poly(A) RNA completely devoid of rRNA. Therefore, measuring the level of a particular modification on mRNA by mass spectrometry must take into account this drawback. Furthermore, potential artefacts can result from antibody cross reactions or partial chemical treatment (e.g., insufficient deamination by bisulfite sequencing can overestimate the abundance of m5C). One way to confirm potential modification sites is to identify the enzymes responsible for their catalysis and perform mapping in knock out conditions. Alternatively, when available, an orthogonal mapping approach could be used for site validation.
m6A on mRNA is mainly installed by a large methyltransferase complex of which METTL3 carries the catalytic activity. In mammals the deposition occurs at the short-degenerated sequence DRACH (D = G/A/U, R = G/A, H = A/U/C), and is enriched near stop codons and on long internal exons.[15-17] A combination of cis-acting elements,[18] epigenetic marks[19] and other transactivating factors[20] helps to ensure the recognition of the target sites by the methyltransferase complex. Other m6A methyltransferases such as METTL16 and METTL4 catalyze m6A on small non-coding RNAs while METTL5 and ZCCHC4 are specific to ribosomal RNAs.[21] In the cytoplasm, m6A plays a preponderant role in mRNA decay[22, 23] and translation,[24] while in the nucleus it can regulate DNA repair,[25] chromatin structure,[26-28] transcription,[29, 30] alternative splicing,[31-34] alternative polyadenylation[35], and mRNA export.[36] The best studied effectors of m6A function, also known as m6A readers, are the members of the YTH protein family, which specifically recognize the modification and trigger the downstream RNA processes.[4, 5] Other identified readers include IGF2BPs and FMR1.[37, 38] Given the widespread role of m6A in mRNA metabolism it is not surprising that its alteration has been linked to numerous developmental and physiological defects in human.[2, 39]
In contrast to m6A, the deamination of adenosine into inosine is catalyzed by a single family of enzymes called ADAR. ADAR proteins preferentially edit strong double-stranded RNA (dsRNA) structures that are present on coding and non-coding RNA, including the inverted Alu repetitive elements.[40-42] The principle of the RNA editing code has been unlocked recently for ADAR1 using a massively parallel synthetic approach.[43] Certain local sequence motifs and minor structural disruption can be favorably edited, which can further propagate the editing events along the dsRNA in a recursive manner. RNA editing is critical to disrupt the structure of endogenous dsRNA and hence to prevent their recognition as foreign nucleic acids by the host immune system.[44, 45] The absence of ADAR1 causes the autoimmune disease Aicardi-Goutières syndrome in children, which result in severe neurological alterations.[46]
The knowledge on individual modifications has expanded rapidly in the last decade owing in part to major improvement in genomic approaches and the motivation to unravel their role in regulating the RNA fate. The specific deposition of certain RNA modifications, as well as their molecular and biological functions have now been thoroughly characterized. While there is still much more to be learned about their function—especially the low abundant ones—several recent reports have suggested an interplay among different RNA marks (Table 1). While this interplay has best been studied for tRNA modifications, some are slowly being uncovered for mRNA modifications. Such interplay does not necessarily imply a close interaction between the marks on same transcripts but any circumstances wherein one mark impacts the level or function of another mark. Here we describe the current methods used to detect RNA modifications and their limitations. We next discuss the potential crosstalk between different RNA modifications, their regulatory players and their participation in similar biological processes. The interplay between tRNA modifications will be only briefly mentioned as this topic has recently been covered in a comprehensive review.[47]
TABLE 1. RNA modifications Possible interplay Model system Unknown aspects/open questions References queosine and m5C Queosinylation at position 34 promotes m5C level at position 38 in tRNAAsp S.pombe, cancer cell lines, mouse Precise mechanismMuller et al. 2015 Nucleic Acids Res
Tuorto et al. 2018 EMBO J
m6A and m5C NSUN2-mediated m5C methylation increases m6A deposition by METTL3-14 and vice versa in the 3’UTR of p21 mRNA Cancer cell lines How joint m6A and m5C enhance p21 translation in oxidative stress-induced cellular senescence Li et al. 2017 J Cell Biochem m6A promotes decay while m5C increases stability of maternal mRNAs during maternal to zygotic transition Zebrafish, mouse What determines the type of methylation on specific transcripts, if m6A and m5C can be present on the same maternal mRNAs and what is the final output on RNA stabilityZhao et al. 2017 Nature
Ivanova et al. 2017 Mol Cell
Sui et al. 2020 Cell Cycle
Yang et al. 2019 Mol Cell
m6A and A-to-I editing Regulation of deposition on mRNA (conflicting data) Human embryonic stem cells, cancer cell lines Precise mechanism and the basis for the differential regulation in different systemsXiang et al. 2018 Mol Cell
Visvanathan et al. 2019 Genes
m6A and Nm LARP7-mediated 2′-O-methylation and METTL16-mediated m6A coexist on U6 snRNA; LARP7 and METTL16 interact in an RNA-dependent way Mouse male germ cells, cancer cell lines Whether the interaction between LARP7 and METTL16 influence 2′-O-methylation/m6A deposition on U6 snRNAWang et al. 2020 Mol Cell
Hasler et al. 2020 Mol Cell
Pendleton et al. 2017 Cell
Shima et al. 2017 Cell Rep
Warda et al. 2017 EMBO Rep
Mendel et al. 2018 Mol Cell
Ishigami et al. 2021 Nat Commun
Reader protein Possible interplay Model system Unknown aspects/open questions References YTHDF proteins (m6A readers) Binding to a m5C-carrying probe (derived from the human CINP gene) Cancer cell lines Binding might occur indirectly via interaction with the m5C reader YBX1 Dai et al. 2020 Anal Chem Binding to a m1A-carrying probe (derived from the human SOX18 gene/28S rRNA or containing m1A within a purine-rich motif) Cancer cell lines m1A is relatively rare on mRNA and the biological relevance of such binding is questionableDai et al. 2018 Anal Chem
Seo et al. 2020 ACS Chem Biol
Zheng et al. 2020 Cell Discov
FMRP (sequence-context-dep m6A reader) Preferential binding to ribosomes carrying specific Nm patterns on rRNA; binding to the BC1 ncRNA in a Nm-dependent way Human embryonic stem cells and neuronal precursor cells, mouse Binding might be indirectD'Souza et al. 2019 iScience
Lacoux et al. 2012 Nucleic Acids Res
Current challenges for the simultaneous detection of multiple RNA modificationsIn the last years, technological advances enabled major improvements in the detection of RNA modifications.[48, 49] Most of the current methods rely either on the particular reverse transcription signatures left by RNA modifications in cDNA, which are naturally occurring or induced by chemical/enzyme-based treatment or to an antibody-based pulldown approach followed by short read sequencing. For a subset of RNA modifications, it is possible to create transcriptome wide maps in a nucleotide-resolution manner and even obtain the stoichiometric quantification of single sites. However, these techniques are limited to detect modifications for which highly specific antibodies or reactive chemical compounds and enzymes are available or so called “hard-stop” modifications, which lead naturally to RT-arrest or other mutation signatures during the reverse transcription.[50] In addition, they often require laborious protocols and most of these approaches enable the detection of only one modification at a time, and therefore rely on the correlation of different datasets to study the interplay among different modifications. Certain methods can be adapted and combined to directly measure several modifications in the same sample,[51] but the rather complex protocols, the need for high amount of input material and the loss of information about their relative distribution to each other make it highly inconvenient. Thus, a need for novel methods to simultaneously measure and identify multiple RNA modifications is imperative. To date, such methods are still in their infancy, but rapid progress in the development of mass spectrometry (MS) approaches, native RNA long read sequencing and nuclear magnetic resonance (NMR) spectroscopy hold great promises.
MS is one of the current approaches used for the determination and quantification of co-occurring RNA modifications.[52, 53] One of the main advantages of the MS-based approach is that it is applicable to all types of modifications. It relies on the property that most modified nucleotides have a unique mass that can be distinguished from each other and with the unmodified counterpart. Therefore, MS allows the analysis of multiple RNA modifications in parallel, including the detection and discovery of previously unknown marks. Nevertheless, the major disadvantage is that crucial information about the location of specific transcripts and the sequence context are lost. A way to overcome these limitations consists in partially digesting the RNA in smaller oligonucleotides to map modified nucleotides to RNA sequences or the direct sequencing of intact, full-length RNAs to compare their mass spectra with sequence databases.[54] This allows to gain insights into the modification landscape of specific RNA at nucleotide resolution. However, this method is not applicable for transcriptome-wide detection and requires the isolation of pure RNA species. More details on these issues can be found in Lauman and Garcia.[53]
An alternative approach is the use of a platform for direct sequencing of RNA molecules without the need for cDNA synthesis or PCR to preserve the information of modified nucleotides. Such native long RNA sequencing is commercially available by Oxford Nanopore Technologies (ONT). In this approach, specific motor proteins actively ensure the transport of a nucleic acid molecule through each pore, which results in a sequence-specific perturbation of the measured current. This change in the current signal can be converted to the corresponding sequence of nucleotides. The presence of RNA modifications can further modify the current and therefore leave a specific footprint. This has been reported for instance for m6A, m5C, 7-Methylguanosine (m7G) and pseudouridine. In certain cases, the modified current is leading to base miscalling and can be recognized as reproducible frameshift, deletion or insertion patterns by adapted base calling methods, as used for A-to-I, m7G and pseudouridine sites.[55-59] Nevertheless, the identification of the current change depends on the sequence context, which means that base calling algorithms should be trained with all possible motifs containing known modifications. In addition, the current change induced by modified nucleotides in comparison to the unmodified counterpart can be very subtle. Therefore, to detect modified sites with high confidence, a knock out condition for the modifying enzyme to measure relative changes of RNA modifications at individual or set of sites can be used.[56, 58] These issues still need to be overcome for most modifications, which is just starting to be explored.
An additionally promising technique to gain more information about the dynamics of RNA modifications is NMR spectroscopy. NMR has been widely used to study the dynamic and structural effects of modification on RNA, however, it can also be used for their identification.[60-66] Recently, a novel time-resolved NMR monitoring of RNA maturation has been proposed.[67] Taking advantage of the non-disruptive nature of NMR, the de novo synthesis of modifications on unmodified RNA has been monitored to study the consecutive generation of RNA modifications on tRNAPhe in a continuous- and time-resolved way. Using this method, a mutual interplay in the generation of Ψ55, m5U54, and m1A58 on tRNAPhe has been identified. While NMR has the advantage to allow a strong assessment of structural features of tRNA at atomic resolution and, therefore, preserve the information about the location of different RNA modifications, it relies on the use of high quantities of isotope labelled RNA. In addition, it is extremely challenging to measure intact, high molecular weight RNAs as so far only a few NMR-based studies could investigate RNA that exceeds a 100-nucleotide length.[68-70] This currently clearly limits the usage of NMR for the investigation of longer RNAs such as mRNA or rRNA.
Interplay in the deposition of RNA modificationsThe understanding of the regulation and function of individual RNA modifications is constantly increasing. Novel enzymes involved in the deposition of different modifications keep being discovered and their mechanisms elucidated. Despite this increasing knowledge about the enzymes essential for the generation of RNA modifications, the mutual influence of RNA modifications remains poorly understood. However, some indications suggest that these influences may in fact not be negligible.
The influence of queuosine on the generation of m5C on tRNA in S. pombe is among the best conserved evidence of a mutual regulation of RNA modifications.[71] Queuosine is a complex modification known to be present on several tRNAs at position 34. It cannot be synthesized de novo by eukaryotes. Therefore, the eukaryotic organism relies on external environmental sources of the queuine base, which is used to synthesize queuosine. Interestingly, the growth of S. pombe cells in the presence of queuine not only increased the queuosine level, but also strongly stimulated the in vivo m5C level at position C38 in tRNAAsp. In the absence of TGT, the specific enzyme responsible for the insertion of queuosine into tRNAs, no increase in the methylation was detectable, indicating that not only the presence, but also the incorporation of queuosine into tRNA is required for m5C deposition. These results were confirmed in mammalian HeLa and human colon carcinoma (HCT116) cells, as well as in vivo by analyzing different tissues of mice fed with a queuine free synthetic diet. A specific decrease of the m5C level at C38 in tRNAAsp, but not in other tRNAs, was observed, which could be restored by the addition of synthetic queuine.[72] While these experiments clearly demonstrate an interplay of these two RNA modifications the precise underlying mechanism still remains to be discovered.
Additional examples of such crosstalk for the deposition of tRNA modifications have been demonstrated, mostly in E. coli and yeast. Importantly most of these crosstalks occurs between modifications present at the anti-codon loop region. Archaea are the exception where several examples of interplay between modifications at the main body of tRNA were detected.[73-76] It is currently unclear whether organisms that live at extreme thermophilic conditions are more dependent on step wise deposition than others, or whether this simply reflects a gap in our understanding in the other organisms.
While the generation of m5C on tRNA can be influenced by queuosine, its presence on mRNA can be determined by another mark. p21 functions as a regulator of cell cycle progression and can act both as tumor suppressor and oncogene.[77]p21 mRNA is modified in its 3′ untranslated region (UTR) by both NSUN2 and METTL3/METTL14 catalyzing m5C and m6A, respectively.[78] In vitro methylation assays using a p21 3′UTR reporter construct in HCT116 cells demonstrated that the pre-methylation by NSUN2 increases m6A deposition by METTL3/METTL14 and vice versa, which ultimately enhances the translation of p21 mRNA. The underlying mechanism of this interplay is currently unclear. Also, whether other mRNAs benefit from the cooperative regulation by m5C and m6A awaits future investigations.
Another interplay involving m6A has recently been suggested with Inosine (I), another abundant modification on mRNA. In human embryonic stem cells (ESCs), the editing level of known A-to-I editing sites was shown to differ between m6A-positive and -negative mRNA populations,[79] being the editing rate higher in the latter. In line with this, depletion of METTL3 in HEK293T and mouse 3T3 cells increased the editing level at certain A-to-I sites, while the knockdown of the m6A eraser FTO caused general A-to-I editing downregulation. By contrast, another study conducted in glioma stem-like cells described an opposing effect wherein a general downregulation of the A-I editing level was observed upon depletion of METTL3, albeit the C-U RNA editing catalyzed by APOBEC was increased.[80] These experiments suggest that these two marks influence the deposition of each other, even though the precise mechanism and the basis for the differential regulation in different systems are currently not understood. Potential models for a direct interplay involve the so-called m6A switch mechanism. The generation of m6A in double-stranded regions, as that is hairpin loops, alters the local RNA secondary structure by destabilization of RNA duplexes. Because ADAR binding relies on the presence of double-stranded RNA regions, the presence of m6A could lead to a loss of ADAR binding sites and, therefore, a modulation of the A-I editing (Figure 1A). In addition, the modulation of the A-I editing by m6A could involve proteins binding specifically to m6A, leading to a sterically block of nearby ADAR binding or A-I editing sites (Figure 1B), or opposingly, the recruitment of ADAR to specific target sites (Figure 1C; examples given below) in the presence of m6A.

Potential mechanisms of the mutual interplay of A-I editing and m6A. The deposition of m6A in double-stranded RNA regions can alter the local structure by destabilization of RNA duplexes (m6A-switch) and, therefore, disfavor the binding of ADAR leading to a suppression of A-I editing (A). A decrease of the A-I editing might also be caused by m6A reader proteins that sterically block Adar binding sites upon binding to m6A (B). Contrary, m6A readers might increase the A-I editing at specific sites by recruiting Adar (C)
The editing level can also be influenced by m6A-dependent expression control of ADARs. The isoform ADAR1p150 is expressed in response to interferons (IFN) to prevent the overactivation of the dsRNA sensing pathway by editing, in order to weaken dsRNA structures.[81] Recently, it was shown that ADAR transcripts are m6A modified and bound by the m6A reader proteins YTHDF1 and YTHDF2.[82] Upon METTL3 and YTHDF1 knockdown, ADAR1p150 showed an attenuated expression in IFNα stimulated cells resulting in an increased innate immune response.[81] Therefore, m6A and YTHDF1 are required to ensure rapid expression response of ADAR1p150 to prevent excessive immune response.
Molecular interplay of reader proteinsThe way modifications dictate the fate of RNA depends on the nature of the chemical adduct. The modification can have a direct impact on the RNA secondary structure, or act as a scaffold for the recruitment of specific functional proteins (reader proteins). Several of these reader proteins have been extensively studied in the context of single RNA modifications and are assumed to bind to only one modification due to specific intrinsic features. Though, several studies identified a couple of reader proteins that show binding to multiple RNA modifications, which could raise some issues about previous interpretations that would need to be addressed in future functional analysis.
The best characterized reader proteins for any RNA modification are the members of the YTH family. In mammals, this protein family consists of five members: YTHDF1/2/3 and YTHDC1/2. They all have in common a YTH domain that can selectively bind to m6A (Figure 2) by the presence of a hydrophobic binding pocket. Interestingly, it was recently suggested that YTH proteins may also bind additional modifications. For instance, YTHDF proteins from HeLa and HEK cells, especially YTHDF2, have been shown to directly bind to a m5C-carrying probe derived from the human CINP gene.[83] Furthermore, the overall level of both m5C and m6A was increased in YTHDF2 pulldown fractions in comparison to the input. Intriguingly, knockout of YTHDF2 resulted in higher m5C levels in rRNA, leading to the alteration of rRNA processing.[83] The precise mechanism for this effect is not yet understood. The binding to m5C requires the same amino acid residue of the YTH domain that was shown to be essential for m6A recognition, suggesting that the interaction with m5C is direct. However, it should be noted that YTHDF1 and YTHDF3 can physically interact with YBX1, a validated m5C reader.[84] This indicates that the binding of YTHDF proteins might also occur indirectly through interaction with this protein (Figure 2).

The binding specificities of YTHDF (DF) proteins. Apart from the binding to m6A, studies indicate the binding of DF2 and DF1/3 to m5C and m1A. The binding of DF1/3 to m5C might be directed by the interaction with the m5C reader protein YBX1. The weight of the arrow indicates the relative strength of the binding
YTHDF proteins were also suggested to bind to the m1A modification (Figure 2). Two independent studies in human cells found an enrichment of YTH proteins after pull down with m1A-containing probes coupled to quantitative proteomics.[85, 86] Further in vitro binding assays with recombinant proteins could validate these associations. It seems that in contrast to m6A binding, only YTHDF proteins can selectively bind m1A while YTHDC1/2 have little affinity for the m1A probe. More recent work found higher enrichment for YTHDF3 in comparison to YTHDF1/2.[87] Such differences might stem from the nature of the m1A probes that were used in each study. Noteworthy, the determined binding affinities of any of the YTH proteins were weaker towards m1A than m6A and m1A is relatively rare on mRNA, therefore, the biological relevance of such binding is questionable.[14] This also hold true for the binding of YTHDFs to m5C. Some correlations were found between m1A-methylated transcripts and YTHDF targets identified by crosslinking and immunoprecipitation (CLIP) experiments but those should be interpreted with caution as m1A antibody can also recognize the cap structure leading to a high rate of false positives.[50, 88]
FMRP is an RNA-binding protein that is essential for brain function. A loss of function of the encoding gene is the main cause of the Fragile X syndrome (FXS) in human, a severe neurodevelopmental disorder characterized by intellectual disability and behavioral alterations.[89] A couple of recent studies suggested that FMRP acts as a m6A reader in order to control the export, stability and translation of methylated RNAs.[37, 90-95] By contrast to YTH proteins, the binding to m6A appears to be sequence-dependent, and may also involve a direct interaction with YTHDF. Interestingly, FMRP was also found in complex with ADAR, the enzyme responsible for A-I editing. This association is conserved in multiple organisms, including Drosophila, zebrafish, mouse and human.[96-99] In all these organisms, the loss of FMRP leads to changes in the editing level. However, the changes can be in both directions and vary between different organisms, suggesting the existence of multiple mechanisms. Proposed models involve the recruitment of ADAR to its mRNA targets through the RNA binding activity of FMRP (Figure 1C), sequestration of ADAR by FMRP for binding to its targets and modulation in ADAR expression upon FMRP loss. It is likely that a combination of these effects occurs depending on the sequence context, cell type and organism. So far, it has not been studied whether these effects are dependent on m6A. As mentioned above, since a correlation between m6A and A-I editing events exist, it will be interesting to investigate if the FMRP control of RNA editing involves its m6A RNA binding ability.
In addition to its implications in the m6A and A-I editing pathways, FMRP has also been shown to affect and read Nm. FMRP interacts with a subset of small nucleolar RNAs (snoRNAs), namely C/D box snoRNAs, that guide the 2'-O-methylation of rRNA.[100] Both the absence and overexpression of FMRP result in a differential 2'-O-methylation pattern of rRNA. Interestingly, the differential rRNA methylation pattern can be recognized by FMRP itself. FMRP preferentially binds ribosomes with hyper-methylated 18S rRNA and hypo-methylated 28S rRNA. Another study showed that FMRP forms a complex with BC1 RNA, a brain specific non-coding RNA involved in translational control, in a 2'-O-methylation manner. In the soma, the presence of Nm on BC1 RNA decreases the binding of FMRP, while at the synapses Nm is virtually absent, enabling the formation of BC1-FMRP-mRNA complexes and the regulation of local translation.[101] Thus, as shown for YTH proteins, FMRP binding to mRNA can be influenced by several modifications.
Currently it is unclear how the modifications impact FMRP binding. In contrast to YTH proteins direct binding of FMRP to m6A or Nm has not been demonstrated. The sequence context seems to be important for FMRP to recognize m6A while Nm may impact the secondary structure and thereby influence FMRP binding. Given the ability of several modifications to alter the secondary structure of the RNA, it is likely that the binding of additional proteins might be impacted by the presence or absence of RNA modifications.
Potential interplay in the control of gene expressionA multitude of different RNA modifications have been identified and the mechanisms by which they individually affect the RNA fate and biological processes, as well as diseases starts to be uncovered. Recent studies reported their involvement in similar molecular processes, yet a potential cooperativity in driving these processes still remain to be formally demonstrated for most of these events. Below we are listing some examples where potential interactions may exist.
TranslationAs mentioned above, one example for a combinatory regulation in the translation process involved the p21 mRNA, which is both m5C- and m6A-modified within its 3′UTR. The combined presence of both types of methylation enhance p21 translation, leading to increased p21 protein levels during cellular senescence induced by oxidative stress.[78] The individual or combined presence of m6A and m5C in the 3′UTR of p21 leads to a similar increase in p21 translation, suggesting that the two methylations could affect translation via the same mechanism. It is unclear how both marks cooperate for this function. The role of YTHDF proteins in translation was recently disputed[23, 102] and no mechanism has been demonstrated yet for the involvement of m5C in translation.
Splicingm6A methylation and A-to-I RNA editing events both happen co-transcriptionally in the nucleus, which implies their potential to regulate any step of the downstream mRNA processing. Several studies revealed crosstalk between the m6A machinery and the regulation of RNA splicing. In Drosophila and human cells, knockdown of individual components of the m6A methyltransferase complex results in alteration of several splicing patterns, such as alternative 5′ss usage and intron retention.[15, 103] The m6A reader protein YTHDC1 was also found to regulate alternative splicing of a subset of m6A modified sites. It can promote exon inclusion by recruiting the splicing enhancer SRSF3 while interfering with the binding of the splicing silencer SRSF10.[104] In addition, m6A erasers partially co-localize with splicing factors in the nucleus and both FTO and ALKBH5 depletion results in alternative splicing defects.[105-107] A-to-I RNA editing and pre-mRNA splicing also influence each other. Loss of ADAR has been shown to induce global changes in splicing in various cell types and model systems.[108-111] A recent genome-wide study in human cells identified around 500 editing sites that could affect splicing by changing the invariant AG dinucleotide to GG in the 3′ss or via ADAR binding to exons located in dsRNA regions.[112] Interestingly, m6A was also shown to be deposited on the 3′ss of the S-adenosylmethionine (SAM) synthetase pre-mRNA in C. elegans by METT-10, the METTL16 ortholog. Methylation at this site prevents the binding of the splicing factor U2AF65 and thereby alters the proper splicing of SAM synthetase.[113, 114] While the sam pre-mRNA is not regulated in this fashion in mammals, the mechanism of splicing inhibition by 3′ss-m6A appears conserved. More studies are needed to decipher to which extent this mechanism participates in splicing regulation in higher eukaryotes and if an interplay with RNA editing occurs. In Drosophila, genome-wide characterization of RNA editing events identified several editing sites located within or in close proximity to alternatively-spliced exons.[115]Sex lethal (Sxl), one of the major regulators of sex determination in flies, is found among the transcripts where RNA editing and alternative splicing might affect each other. Interestingly, m6A modification is known to be required for proper splicing of the Sxl transcript and m6A writer mutants display decreased levels of the female-specific isoform together with the appearance of the male-specific exon in female flies.[32, 103, 116] In light of this, it would be interesting to investigate how splicing is affected when different modifications are present on the same mRNA and whether a “splicing code” based on RNA modifications could contribute to fine-tune the splicing of modified transcripts.
Apart from the influence of modifications on the mRNA splicing pattern, modifications on the spliceosomal RNA also participate to splicing regulation. A recent study revealed that the La-related protein LARP7 interacts with the box C/D snoRNP facilitating U6 2′-O-methylation in mouse male germ cells.[117] U6 snRNA is an essential catalytic component of the spliceosome and defective ribose methylation in the absence of LARP7 leads to severe changes in pre-mRNA splicing, including skipped exons, retained introns as well as alternative 5′ and 3′ss. Similar findings were reported also in human HEK293
留言 (0)