

記住我
For all of their complexity and rich diversity of constituent cellular phenotypes, multicellular organisms can be characterized by a common foundation—their genome. With all of our cells sharing the same genetic code, regulation of gene expression serves as the root of heterogeneity in cellular identity, response, and role. Given all of the information (form, function, development from a single cell, etc.) that must be encoded in the human genome, it is perhaps no surprise that the diploid human genome is very long, spanning 6 billion base pairs. Stretched end-to-end, the DNA in each diploid human somatic cell would measure roughly 2 m; however, a need for space-efficient storage of DNA results in its compaction by orders-of-magnitude to fit inside small nuclei less than 10 μm in diameter (Greeley, Crapo, & Vollmer, 1978; Piovesan et al., 2019). Despite this compaction, DNA must also be dynamically accessible to allow gene activation, regulation, and replication as the cell grows, divides, and responds to stimuli. These considerations define the two seemingly contradictory challenges of chromatin organization: packaging DNA so that it fits within the cell while retaining sufficient accessibility for processes necessary for cell functionality.
Looking for the forces managing the balance of packaging versus functional accessibility, researchers dove into an exploration of the linear genome in the 1970s. The recombinant DNA revolution heralded the development of new experimental techniques for molecular genetics (e.g., isolating genes for study), and genes were sequenced for the first time (Lis, 2019). By the 1980s and 90s, scientists had uncovered a myriad of factors involved with transcriptional initiation and regulation at regulatory motifs proximal to the gene of interest (Lambert et al., 2018; Roeder, 2019). As our understanding of the complexity of transcriptional regulation deepened, however, it became apparent that proximal regulatory elements are just one part of a wider regulatory landscape. The subject of the 3D genome and distal regulation of transcription began capturing greater interest as researchers identified regulatory elements termed enhancers thousands to millions of base pairs distal to their target genes (Spitz, 2016). For instance, a nuclear ligation assay developed in 1993 probed the rat prolactin gene and reported that the distal enhancer and proximal promoter regions are spatially juxtaposed, an interaction stimulated by estrogen ligand acting upon an estrogen receptor bound to the distal enhancer (Cullen, Kladde, & Seyfred, 1993; Gothard, Hibbard, & Seyfred, 1996). Given the central role that gene expression plays in cell phenotype and the onset of disease, unpacking the functional ramifications of these distal genetic interactions holds great promise for advancing our understanding of the genome and has thus become the impetus for the development of chromosome conformation capture technologies. In this review, we chronicle major developments in chromosome conformation capture technology and the biological insights their application has given us, with particular attention given to the recently developed Micro-C method.
2 THE DEVELOPMENT OF CHROMOSOME CONFORMATION CAPTURE TECHNOLOGIES 2.1 Chronology of key technologies and the features they detect3C, 4C, and 5C—Chromosome conformation capture technologies have primarily derived from the foundation laid by 3C (Chromosome Conformation Capture) in 2002 (Figure 1a; Dekker, Rippe, Dekker, & Kleckner, 2002). First developed in yeast (Dekker et al., 2002) and soon adapted for mammalian cells (Tolhuis, Palstra, Splinter, Grosveld, & de Laat, 2002), 3C is capable of probing pairwise interactions between specific genetic loci of interest and generating population-averaged contact frequency estimates between two chromosomal loci. Moreover, applying 3C to estimate the pairwise interaction frequencies of multiple loci enables the development of experimentally-constrained 3D polymer models of chromosomes (Dekker et al., 2002). The unique ability of 3C to precisely focus on specific loci and generate such models at relatively high resolution overcame some of the limitations of microscopic methods such as electron microscopy (which lacks locus specificity) and fluorescence in situ hybridization (FISH, which is a lower throughput technique) previously harnessed in probing nuclear architecture (Barutcu et al., 2016; Belmont, 2014; Sati & Cavalli, 2017). The steps at the heart of the 3C protocol—namely, chemical fixation and cross-linking of DNA, restriction enzyme (RE) digestion, proximity ligation, cross-linking reversal, and PCR amplification to generate the interactome library—allow 3C to achieve its specificity (by virtue of locus-specific amplification) and throughput (by virtue of its scalability to the whole-genome scale) and have since become a mainstay of the 3D genome field.

Timeline and comparison of major chromosome conformation capture techniques. (a) Chronological development of chromosome conformation capture technologies colored by type of method. Major observational, mechanistic, or biological discoveries are listed above the timeline. (b) Comparison of landmark 3C-based methods and the resolutions at which their datasets are typically analyzed. The typical resolution ranges for these technologies are historically grounded and may widen or shift with the inclusion of recent advances in methodology. Resolutions at which key features of chromatin organization typically manifest are shown on the right
However, 3C only probes interactions between pairs of loci for which PCR primers have been designed, making it a low-throughput technique that is normally analyzed on gels or with RT-qPCR. This limitation as a “one versus one” method (Figure 1b) prompted the development of two higher-throughput derivatives, Circular Chromosome Conformation Capture or Chromosome Conformation Capture-on-Chip (4C) and Chromosome Conformation Capture Carbon Copy (5C), in 2006 (Dostie et al., 2006; Simonis et al., 2006; Z. Zhao et al., 2006). 4C modifies the 3C protocol by circularizing ligated fragments, allowing inverse PCR amplification that only requires primers for one of any two ligated fragments; thus, 4C is capable of mapping the interactions between a known locus of interest and the entire genome (a “one versus all” method, Figure 1b; Simonis et al., 2006; Z. Zhao et al., 2006). In its initial application, 4C examined the H19 imprinting control region and revealed that direct long-range interaction between methylated regions can serve as an epigenetic regulatory mechanism for transcription (Z. Zhao et al., 2006). By contrast, 5C amplifies select parts of a 3C library by using PCR primer pairs to focus on a region of interest for analysis via sequencing or microarrays (Dostie et al., 2006). Accordingly, 5C is able to probe pairwise interactions across a whole region of interest (a “many versus many” method, Figure 1b) and revealed looping interactions within the genome, affirming on a broader scale prior 3C studies on looping in the β-globin locus (Dostie et al., 2006; Tolhuis et al., 2002; Vakoc et al., 2005). Subsequently, researchers studying transcriptional regulation of the X-inactivation center in mouse embryonic stem cells (mESCs) using 5C and FISH discovered the presence of 200 kb–1 Mb sized self-interacting DNA regions they termed topologically associating domains (TADs; Nora et al., 2012). TADs define local regions of the genome that preferentially self-interact at a significantly increased frequency (typically ~2–3 fold greater) relative to regions outside of the TAD (Chang, Ghosh, & Noordermeer, 2020; Dixon et al., 2012; Nora et al., 2012; Rao et al., 2014). TADs are demarcated by clearly defined boundaries and can nest within compartments or within one another as smaller “subTADs” manifest within larger TADs. Despite the ubiquity of TADs as features of the 3D genome, it is important to note that the field currently lacks a unified definition for what constitutes a TAD and uncertainty remains in terms of nomenclature (Box ; Beagan & Phillips-Cremins, 2020; Rowley & Corces, 2018). Despite their description less than a decade ago, TADs are now recognized as a hallmark of chromatin organization at the scale of tens of kilobases to megabases.
Hi-C—Although 3C, 4C, and 5C allowed long-range DNA interactions to be studied, their reliance on researchers choosing target loci of interest prevented them from probing the whole genome in an unbiased manner. The advent of high-throughput chromosome conformation capture (Hi-C) in 2009 altered this paradigm (Lieberman-Aiden et al., 2009). A genome-wide adaptation of 3C, Hi-C utilizes biotinylation to enrich for proximity ligated contacts and modifies the library amplification process to utilize universal adapters and primers for high-throughput sequencing. Agnostic to the specific sequences being amplified, Hi-C can thus probe all genomic interactions in an unbiased “all versus all” approach (Figure 1b). The original Hi-C protocol, also referred to as dilution Hi-C, uses dilute proximity ligation conditions to minimize artifacts following nuclear lysis; this methodology was altered in the development of subsequent Hi-C derivatives. First tested in a human lymphoblastoid line, the initial application of low-resolution Hi-C (1 Mb-level resolution, achieved with 8.4 million reads) revealed the presence of preferentially self-interacting A and B compartments as a novel level of genome organization (Figure 2; Lieberman-Aiden et al., 2009). “A” compartments largely contain DNA classified as euchromatin that is more transcriptionally active, less densely packed, and localized away from the nuclear periphery, with the notable exception of nuclear pore complexes (Gozalo et al., 2020; Hildebrand & Dekker, 2020). By contrast, “B” compartments largely contain DNA classified as heterochromatin that is transcriptionally inactive, more densely packed, and localized near the nuclear periphery or the nucleolus (Hildebrand & Dekker, 2020). Manifesting genome-wide at the megabase scale and capable of checkering across chromosomes (unlike TADs), compartments exhibit a significantly higher frequency of long-range inter- and intrachromosomal interactions with DNA in the same compartment type compared to DNA in the alternative compartment type (Lieberman-Aiden et al., 2009) and correlate strongly with patterns in the timing of DNA replication (called replication timing domains) observed by BrdU labeling and Repli-seq (Hiratani et al., 2008; Rhind & Gilbert, 2013; P. A. Zhao, Sasaki, & Gilbert, 2020).
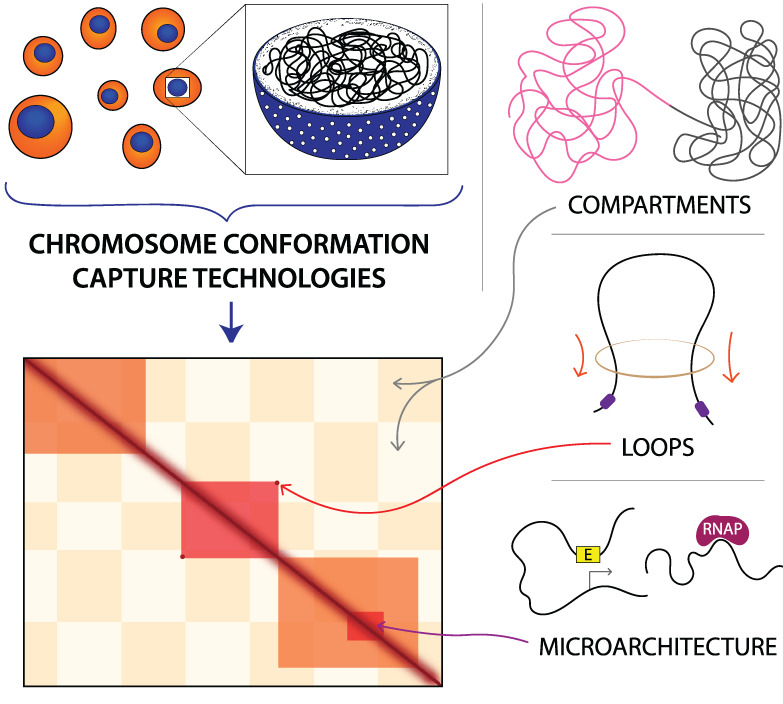
 Micro-C captures finer-scale features of chromatin organization than Hi-C. (Top row) High-resolution Hi-C (Bonev et al., 2017) and Micro-C (Hsieh et al., 2020) datasets generated from wild-type mESCs are visually juxtaposed at various scales of chromosomal organization. The data is binned at different resolutions using HiGlass, with the visualization of any particular feature requiring bins of a finer resolution than the size of the feature. Contact heatmaps of the whole genome, compartments, topologically associating domains (TADs), and loops are shown. The checkerboard pattern in the second column of plots indicates separation into A/B compartments. The markers in the third column of plots indicate TADs, while the markers in the fourth column of plots indicate corner peaks or “dots” specifying loops. (Bottom row) Fine-scale resolution maps of the same Hi-C and Micro-C datasets. Markers identify microarchitecture, such as enhancer-promoter (E–P) or promoter-promoter (P–P) loops, stripes, and domains, visible in Micro-C but not discernable in Hi-C, and genes within the region are annotated below. This figure is inspired by fig. 1d and S1d in Hsieh et al. (2020)
Micro-C captures finer-scale features of chromatin organization than Hi-C. (Top row) High-resolution Hi-C (Bonev et al., 2017) and Micro-C (Hsieh et al., 2020) datasets generated from wild-type mESCs are visually juxtaposed at various scales of chromosomal organization. The data is binned at different resolutions using HiGlass, with the visualization of any particular feature requiring bins of a finer resolution than the size of the feature. Contact heatmaps of the whole genome, compartments, topologically associating domains (TADs), and loops are shown. The checkerboard pattern in the second column of plots indicates separation into A/B compartments. The markers in the third column of plots indicate TADs, while the markers in the fourth column of plots indicate corner peaks or “dots” specifying loops. (Bottom row) Fine-scale resolution maps of the same Hi-C and Micro-C datasets. Markers identify microarchitecture, such as enhancer-promoter (E–P) or promoter-promoter (P–P) loops, stripes, and domains, visible in Micro-C but not discernable in Hi-C, and genes within the region are annotated below. This figure is inspired by fig. 1d and S1d in Hsieh et al. (2020)
Equipped with a genome-wide method for conformation capture, researchers began exploring sub-megabase levels of nuclear architecture across the genome. The opening of this sub-megabase frontier was intimately linked to advances in DNA sequencing, with costs dropping faster than Moore's law from 2008 onward due to the advent of next-generation sequencing (Heather & Chain, 2016; Wetterstrand, 2020). Innovation in sequencing was a necessary companion to Hi-C because, unlike nonpairwise genomic sequencing methods (e.g., RNA-seq, ChIP-seq) whose required sequencing reads for a resolution n scales linearly with genomic size, increasing the resolution of a genome-wide Hi-C pairwise contact matrix n-fold necessitates n2 reads. Thus, sequencing costs serve as a limiting factor for the depth at which the interactome is captured.
Early Hi-C analysis of genomes in hESCs, mESCs, and differentiated cell types identified TADs as genome-wide features of mammalian nuclear architecture and reported that TADs are largely invariant between cell types, evolutionarily conserved, and separated by boundary regions enriched for factors and housekeeping genes of interest (Dixon et al., 2012; Phillips-Cremins et al., 2013). Subsequently, the first high-resolution contact maps generated by the application of in situ Hi-C in human and mouse cell lines revealed levels of genome organization as fine as the 1-kb scale from ~5 billion sequencing reads (Rao et al., 2014). Dots (corner peaks) were now clearly visible in Hi-C maps, and domains with clear corner peaks were termed loop domains (Figure 2). By comparing with ChIP-seq analyses, Rao et al. reported that 86% of these loop domains are bound by the CCCTC-binding factor (CTCF), 92% of which demarcate loop boundaries in a convergent orientation, and that 86–87% are bound by cohesin (a Structural Maintenance of Chromosomes, or SMC, complex) subunits RAD21 and SMC3 (Rao et al., 2014). Of the 2,854 loops identified as involving enhancers and promoters (E–P loops), 557 were cell-type-specific and also strongly correlated with cell-type-specific gene activation, thus ascribing a more definitively functional role to contact map features (Rao et al., 2014). Further probing the relationship between nuclear architecture, gene expression, and cell fate, the first high-resolution Hi-C analysis of development mapped 3D genome organization during mouse neural differentiation in vitro and in vivo (Bonev et al., 2017). Examination of the data revealed that transcriptional changes during differentiation are correlated with alterations in the strength of long-range interactions and the emergence of cell type-specific enhancer–promoter (E–P) contacts. Bonev et al. also found that such E–P interactions occur primarily within the same TAD and are generally established alongside gene expression, affirming similar findings of how TADs constrain enhancer activity (Symmons et al., 2014) and further connecting form and function. Critically, insights into the extensive genomic rewiring of structure during development underscored the dynamism of nuclear architecture and helped shift the field from a fairly cell-type invariant view of chromatin spatial organization toward a more cell-type-specific one (Beagan et al., 2017; Bonev et al., 2017; Pękowska et al., 2018). A comparison and timeline of key chromosome conformation capture methods are shown in Figure 1, and a visualization of Hi-C map features is shown in Figure 2.
2.2 Other 3C-derived technologiesAs a rapidly blossoming field, nuclear architecture has witnessed an explosion in technology development as 3C-based methods have been modified to create a diverse array of derivatives. Many of these derivatives are designed to address the shortcomings and limitations of their parent technologies; others act to incorporate breakthroughs in adjacent fields. Some common modifications to parent protocols are reflected by a sampling of noteworthy 3C, 4C, and 5C derivatives. In an effort to bypass potential biases introduced by chemical cross-linking, intrinsic 3C (i3C), and 4C (i4C) forgo cross-linking and perform digestion and ligation in situ (discussed below); this not only reconstitutes known features of folding, but also improves the signal from more stable chromatin interactions (Brant et al., 2016). 4C-seq improves 4C's resolution and reproducibility by introducing a second round of restriction digestion and ligation, and improves 4C's throughput by incorporating adapters for NGS (van de Werken et al., 2012). The addition of unique molecular identifiers (UMIs) to 4C-seq in UMI-4C further refines the protocol, improving sensitivity, specificity, and multiplexing (Schwartzman et al., 2016). Finally, 5C-ID performs ligation-mediated amplification with a double alternating primer design and uses in situ digestion and ligation, resulting in reduced noise, improved sensitivity to loops, and fewer required input cells than native 5C (J. H. Kim et al., 2018). Major categories of 3C-based derivatives—namely, in situ and single-cell Hi-C, ChIP-based methods, and capture-based methods—are briefly discussed here.
In situ and single-cell Hi-C—In its initial development, single-cell Hi-C (scHi-C) adopted an approach employed by the nuclear ligation assay by performing cross-linking, restriction digestion, and ligation within intact nuclei, after which it isolated individual nuclei and proceeded through the rest of the Hi-C methodology (Cullen et al., 1993; Nagano et al., 2013). In situ bulk Hi-C subsequently drew upon similar inspiration by revising the original Hi-C protocol to perform ligation within permeabilized intact nuclei (Rao et al., 2014). These protocol adjustments enable ligation in smaller volumes, reduce the frequency of spurious contacts, and improve digestion efficiency, resulting in cleaner and higher-resolution data (Nagano, Várnai, et al., 2015; Rao et al., 2014). Subsequent scHi-C derivatives, such as single-nucleus Hi-C (snHi-C) and single-cell combinatorial indexed Hi-C (sciHi-C), have generated multiplexed libraries using tagmentation and indexing (Ramani et al., 2017), improved nuclear sorting efficiency using FACS (Nagano et al., 2017), or minimized contact loss using in situ whole-genome amplification (Flyamer et al., 2017). Given the population-averaged nature of genomic interactome data from 3C-based methods, scHi-C has proven instrumental in distilling cell-to-cell structural heterogeneity, identifying rare cellular subpopulations, and understanding how different levels of organization interact (Nagano et al., 2013; Stevens et al., 2017; Ulianov, Tachibana-Konwalski, & Razin, 2017). For example, Nagano et al. found that transcriptionally active domains hundreds of kb to megabases in size localize to the peripheries of territories hundreds of Mb in size, and Stevens et al. reported that while TADs and loops substantially vary from cell-to-cell, compartments, lamina-associated domains, and active enhancers and promoters do not. Notably, scHi-C has also disentangled cell-cycle dynamics governing features of 3D nuclear organization (Nagano et al., 2017) and revealed developmentally-linked chromatin reorganization in the oocyte-to-zygote transition, such as the presence of TADs and loops but not compartments in zygotic maternal chromatin (Flyamer et al., 2017). However, scHi-C is limited in its ability to detect any given contact because it only captures data from a cell's one (zygotic studies) or two (somatic studies) alleles for any given locus and because the likelihood of detecting an interaction is low (Nagano, Lubling, et al., 2015; Tan, Xing, Chang, Li, & Xie, 2018). Thus, scHi-C faces challenges in separating technical noise from biological variation, and the sparsity of the contact matrix for any given cell presents a challenge in data analysis and interpretation (Lähnemann et al., 2020; Ulianov et al., 2017; Zhu & Wang, 2019).
ChIP-based methods—Interest in the protein-DNA interactions contributing to the 3D genome has spawned the inclusion of chromatin immunoprecipitation (ChIP) methodology into chromosome conformation capture protocols. First introduced in 2005, ChIP-loop (also known as ChIP-3C) modifies the 3C protocol by enriching ligated fragments for contact with a protein of interest (Horike, Cai, Miyano, Cheng, & Kohwi-Shigematsu, 2005). As a “one versus one” method (Figure 1b), ChIP-loop was initially used to investigate chromatin interactions bound by MeCP2, SATB1, or ERα (S. Cai, Lee, & Kohwi-Shigematsu, 2006; Carroll et al., 2005; Fullwood & Ruan, 2009; Horike et al., 2005). Substituting in cloning-based contact analysis in lieu of PCR amplification yields 6C (combined 3C-ChIP-cloning), a method first used to examine the role of EZH2 in mediating long-range contacts (Tiwari, Cope, McGarvey, Ohm, & Baylin, 2008). However, difficulty quantifying ChIP enrichment of inherently noisy 3C data made distinguishing specific interactions from nonspecific false positives in ChIP-loop-based techniques challenging (Fullwood & Ruan, 2009). The whole-genome (“all versus all,” Figure 1b) adaption of ChIP-loop methods, called chromatin interaction analysis by paired-end tag sequencing (ChIA-PET), increases the signal-to-noise ratio (SNR) by using sonication to fragment DNA, adds linker sequences between ligated fragments for ease of extraction, and utilizes adapters for high-throughput paired-end sequencing (Fullwood et al., 2009). Examples of early applications of ChIA-PET include investigations of the chromatin interactomes of ERα (Fullwood et al., 2009) and CTCF (Handoko et al., 2011).
By virtue of their enrichment of protein-centered chromatin interactions, genome-wide ChIP-based conformation capture methods are capable of recapitulating key features of the 3D genome (e.g., TADs and loops) and achieving finer resolution than Hi-C (e.g., by enriching for determinants of organization, such as CTCF and cohesin). Given similarities in methodology, subsequent advances in whole-genome ChIP-based techniques have often drawn from parallel advancements in Hi-C-based techniques. For example, HiChIP, which exhibits improved sensitivity to DNA contacts and lowered input material requirements relative to ChIA-PET, was built by leveraging principles of in situ Hi-C (Mumbach et al., 2016). More recently, multiplex chromatin-interaction analysis via droplet-based and barcode-linked sequencing (ChIA-Drop) has been developed by utilizing microfluidics to partition cross-linked chromatin complexes into gel-bead-in-emulsion (GEM) droplets for subsequent barcoding, amplification, and sequencing (Zheng et al., 2019). Able to probe chromatin interactions with greater precision and resolution than ChIA-PET, ChIP-Drop has been used to explore transcriptionally relevant promoter-centered interactions and shows promise for uncovering novel single-molecule-resolution multi-way contacts (Kempfer & Pombo, 2020; Zheng et al., 2019).
Capture-based methods—With 3C-based methods at the time unable to map cis interactions at a sufficiently high resolution to capture enhancer-promoter contacts in a high-throughput manner, Capture-C was developed in 2014 as a new approach to exploring cis regulation (Hughes et al., 2014). Combining 3C, oligonucleotide capture technology (OCT), and high-throughput sequencing, Capture-C utilizes RNA biotinylated oligo probes to enrich for DNA fragments containing viewpoints of interest. As such, Capture-C can be applied to probe a specific contiguous region (a “many versus many” experiment similar to 5C) or, if probes are designed for, for example, all promoters, Capture-C can serve as a massively parallel 4C (“many versus all”) experiment (Figure 1b). The initial application of Capture-C to the promoters of genes of interest demonstrated the method's ability to elucidate general principles of cis regulation and link single-nucleotide polymorphisms (SNPs) in distal elements to expression changes in their cognate genes (Hughes et al., 2014). Soon after, similar target sequence enrichment was applied to Hi-C libraries to yield Capture Hi-C (CHi-C), further facilitating the discovery of novel long-range promoter contacts (Jäger et al., 2015; Mifsud et al., 2015). Both Capture-C and Capture Hi-C have been applied to investigate gene loci implicated in limb development, demonstrating the phenotypic implications of disrupting enhancer-promoter chromatin structure and uncovering two regimes of chromatin folding: one associated with CTCF/RAD21 binding that is stable across tissues, and another that correlates with tissue-specific changes in chromatin microarchitecture (Andrey et al., 2017; Paliou et al., 2019). Similarly, Capture Hi-C and 4C-seq testing of intra-TAD and inter-TAD insulation in response to genomic duplications revealed the formation of new chromatin domains with pathogenic consequences, further linking structure to phenotype (Franke et al., 2016).
Noting that Capture-C's reliance upon oligonucleotides synthesized on microarrays yielded high costs per sample for small experimental designs and that the Capture-C method was not sensitive enough to detect weak interactions, researchers developed next-generation (NG) Capture-C as a solution (Davies et al., 2016). The NG Capture-C method performs capture using DNA (rather than RNA) biotinylated oligos, introduces a second round of capture, and pools multiple independent 3C libraries for processing in a singular reaction, thus improving sensitivity and throughput. The latest iterations of Capture-C design, Nuclear-Titrated (NuTi) Capture-C and Tiled-C, further advance the resolution and efficacy of Capture-C-based methods (Downes et al., 2020; Oudelaar et al., 2020). NuTi Capture-C isolates 3C libraries from intact nuclei by separating 3C libraries into nuclear and non-nuclear fractions post-ligation and utilizes shorter oligonucleotide probes (shortened from 120 to 50 bp). Tiled-C uses a panel of capture oligonucleotides tiled across all restriction fragments of a region of interest; combined with an optimized protocol which minimizes losses and maximizes library complexity, this allows the method to generate high-resolution contact maps from inputs of very few cells (as low as 2,000). In its initial application, Tiled-C followed the nuclear architecture of mouse erythroid genes of interest through in vivo erythroid differentiation and revealed that structural reorganization within TADs and the emergence of E–P contacts occurs during differentiation, suggesting that chromatin architecture and gene activation are linked (Oudelaar et al., 2020).
Traditional 3C-based methods are limited to detecting pairwise contacts, preventing them from determining the interdependence of multi-way chromatin contacts commonly found in biological systems (Kempfer & Pombo, 2020). For instance, if loci A and B both interact with locus C, do they do so in a mutually exclusive, mutually dependent, or independent manner? 3C-based methods such as chromosomal walks (Olivares-Chauvet et al., 2016) and the concatemer ligation assay (Darrow et al., 2016) began addressing this question by 2016 and, looking to probe multi-way contacts with single-allele resolution, researchers created Tri-C in 2018 (Oudelaar et al., 2018). Employing sonication of ligated fragments to ~450 bp in size, Tri-C creates libraries where a majority of fragments contain multiple ligation junctions that can then be captured by OCT, PCR amplified, and sequenced. Examination of domains containing mouse globin loci using Tri-C reveals regulatory hubs containing multiple enhancers and promoters, as well as heterogeneous patterns of CTCF interaction indicating highly variable chromatin domain formation (Oudelaar et al., 2018). Similarly, multi-contact 4C (MC-4C), published concurrently with Tri-C, has been used to analyze three-way contacts within a region of interest and disentangle cooperative interactions from competitive or random interactions (Allahyar et al., 2018). Recently, the combination of ATAC-seq (Assay for Transposase-Accessible Chromatin using sequencing), i4C-seq, CRISPR/Cas9 modification, and single-molecule RNA FISH to study mechanisms of action underlying cytokine-activated enhancer activity has yielded a cross-linking-free variant of MC-4C and allowed multi-way interactions spanning TAD boundaries to be studied (Weiterer et al., 2020).
2.3 Non 3C-based technologies to map the 3D genomeSPRITE and GAM—While the exploration of the 3D genome has largely been propelled by 3C-derived technologies, other 3D mapping methods have revealed unique insights about genome organization by capturing interactions not preserved by 3C-based methodologies. For example, despite being the gold standard in chromosome conformation capture, Hi-C is limited in its reliance on restriction digestion and proximity ligation; restriction digestion imposes a limit on the resolution of captured data that can be achieved, while proximity ligation biases analysis to primarily pair-wise interactions, may be inefficient, and can invite the inclusion of spurious contacts. Developed as a method for investigating higher-order organizational interactions not captured by proximity ligation, split-pool recognition of interactions by tag extension (SPRITE) addresses some of Hi-C's shortcomings and reveals hubs of interaction within the nucleus (Quinodoz et al., 2018). In a nutshell, the SPRITE method cross-links DNA, RNA, and proteins within a cell, isolates the nucleus, fragments chromatin, sequentially barcodes interacting molecules within each individual complex through several rounds of split-pooling, and exhaustively sequences the barcoded molecules to reconstruct the interactome. By focusing on hubs of cross-linked interactions and forgoing both restriction digestion and proximity ligation, SPRITE is able to probe multi-way contacts across a wide range of nuclear distances at high resolution. Free from a resolution ceiling imposed by RE digestion, SPRITE performs chromatin fragmentation using sonication and DNase digestion; in its initial application, this digestion was optimized to produce DNA fragments 150–1,000 bp in length (Quinodoz et al., 2018). With ~1.5 billion sequencing reads, SPRITE achieved similar kilobase-scale resolution as high-resolution Hi-C and recapitulated known structures of nuclear architecture. Using SPRITE, Quinodoz et al. also observed long-range nuclear interactions in which gene-dense and highly transcribed regions preferentially localize around nuclear speckles while gene-poor and transcriptionally inactive regions localize around the nucleolus. In addition to its ability to detect higher-order genomic organization beyond pairwise interactions, SPRITE can simultaneously explore both the DNA and RNA interactomes, requires fewer input cells than Hi-C, and captures long-range interactions involving actively transcribed enhancers and promoters rarely seen in Hi-C data; conversely, however, it is also a more laborious, lower-throughput process than Hi-C and the efficiency of serially ligating small oligonucleotides for barcoding remains uncharacterized (Fiorillo et al., 2020; Kempfer & Pombo, 2020).
Another technique, Genome Architecture Mapping (GAM), overcomes the limited ability of 3C-based methods to capture multi-way simultaneous interactions (e.g., triplet contacts) and other aspects of organization such as compaction and association with the nuclear periphery (Beagrie et al., 2017). GAM measures chromatin contacts by thinly cryo-sectioning fixed cells, isolating nuclear profiles, and extracting, amplifying, and sequencing the DNA within many such randomly sliced profiles to map the co-segregation rates of all possible pairs of genomic loci using the SLICE (statistical inference of co-segregation) analysis method. The number of collected nuclear slices determines the resolution of GAM data sets, with 400 nuclear slices sequenced with ~1 million reads per slice yielding comparable pairwise contact resolution (30 kb) to Hi-C (Kempfer & Pombo, 2020). As no maximum resolution limit has been achieved for GAM, larger datasets should yield finer resolution data in the future. Successfully benchmarked against Hi-C maps, GAM has shown that enhancers and active genes are enriched among specifically interacting genomic regions, with particularly strong enrichment at transcriptional start and end sites (TSSs & TESs; Beagrie et al., 2017). When compared to Hi-C, GAM is advantageous for its lack of RE digestion, proximity ligation, or chromatin fragmentation and exhibits a superior robustness to noise at large genomic distances, requires fewer input cells, and is well-suited for analysis of complex tissues (e.g., patient biopsies; Beagrie et al., 2017; Fiorillo et al., 2020). Moreover, like SPRITE, GAM's detection of long-range contacts of transcriptionally active enhancers and promoters outpaces Hi-C, as such contacts are difficult to discern in Hi-C data. However, GAM is limited by its dependence upon specialized equipment and training (e.g., for fine cryo-sectioning) and increased complexity of data interpretation (Kempfer & Pombo, 2020).
Though not a measure of chromosome conformation (and accordingly not discussed here in depth), ionizing radiation-induced spatially correlated cleavage of DNA with sequencing (RICC-seq) is another noteworthy technique that has revealed nucleosome-level folding and local structure (Risca, Denny, Straight, & Greenleaf, 2017). Alternatively, the development of i3C, accompanied by the TALE-iD method for validating i3C and i4C interactions via TAL-effector-directed methylation of target enhancers, provided an early framework for cross-linking-free chromosome conformation capture (Brant et al., 2016). This approach was recently further developed in the combination of DNA adenine methyltransferase identification (DamID) with chromosome conformation capture in the DamC method, making it possible to study 3D genome architecture without cross-linking and proximity ligation (Redolfi et al., 2019).
RNA-based methods—Though long the primary focus in nuclear architecture, the DNA interactome is one structural piece among many in the nucleus. In recent years, advances in techniques probing DNA–DNA and DNA-protein contacts have been adapted to study the DNA–RNA and RNA–RNA interactomes. For the sake of brevity, we touch upon only a few “all versus all” technologies in this area. Methods for studying DNA–RNA interactions, such as chromatin-associated RNA sequencing (ChAR-seq) (J. C. Bell et al., 2018), global RNA interactions with DNA by deep sequencing (GRID-seq) (Li et al., 2017), and mapping RNA-genome interactions (MARGI) (Sridhar et al., 2017; Wu et al., 2019) follow the same core workflow: cross-link cells, ligate a linker to cross-linked RNA, reverse transcribe, proximity ligate the cDNA-linker to DNA, and isolate ligated pairs for amplification and paired-end sequencing. Differences between these techniques arise in the design of the linker or “bridge” and the ordering of steps, but all have been instrumental in identifying novel transcript-DNA interactions and distilling the contacts structurally underpinning transcription. RNA–RNA methods, such as RNA hybrid and individual-nucleotide resolution ultraviolet cross-linking and immunoprecipitation (hiCLIP) (Sugimoto et al., 2015) and RNA in situ conformation sequencing (RIC-seq) (Z. Cai et al., 2020), fix and cross-link cells and often utilize proximity ligation to link RNA–RNA-contacts mediated by RNA-binding proteins (RBPs). With RIC-seq achieving single-nucleotide resolution and revealing the necessary role of ncRNA in looping at the MYC and PVT1 loci (Z. Cai et al., 2020), we anticipate the rapid discovery of more RNA-dependent nuclear organization in coming years.
Microscopy-based methods—Beyond the sequencing-based methods discussed above, microscopy-based methods have been instrumental in visualizing nuclear architecture, measuring characteristics of folding (e.g., compaction), identifying chromatin domains, and distilling cellular heterogeneity. With advancements in super-resolution microscopy and synthetic oligonucleotide design, techniques such as fluorescence in situ hybridization (FISH), OligoPAINT, three-dimensional assay for transposase-accessible chromatin-photoactivated localization microscopy (3D ATAC-PALM), stochastic optical reconstruction microscopy (STORM), optical reconstruction of chromatin architecture (ORCA), and live-cell imaging have uncovered structural insights and multiway interactions not captured by the other methods discussed here (Alexander et al., 2019; Beliveau et al., 2012; Chen et al., 2018; Marbouty et al., 2015; Mateo et al., 2019; Rust, Bates, & Zhuang, 2006; Xie et al., 2020). For the sake of brevity, we do not discuss these microscopy-based methods in detail here and instead refer the reader to excellent reviews on the topic (Boettiger & Murphy, 2020; Cattoni, Valeri, Le Gall, & Nollmann, 2015; Kempfer & Pombo, 2020; Lakadamyali & Cosma, 2020).
3 MECHANISTIC UNDERPINNINGS OF NUCLEAR ARCHITECTUREThough the development of 3C-based technologies uncovered features of chromatin organization, understanding the biological mechanisms underlying the features necessitated further experimentation. The past half-decade in particular has seen a renaissance in our mechanistic understanding of 3D genome organizational features as researchers have unearthed the roles of key proteins, forces, and mechanisms and developed advanced models explaining DNA conformation. Specifically, landmark discoveries that have shaped the field's current biological grounding involve the elucidation of the roles of CTCF and cohesin, the loop extrusion model, compartmentalization, and the interplay of different forces across levels of organization.
3.1 The roles of CTCF and cohesinAlthough the development of Hi-C established TADs as hallmarks of genome organization, uncertainty remained over how TADs are formed, maintained, and regulated. Interest in mechanisms capable of locally confining genomic regions from one another began drawing upon decades of research into transcriptional regulation. Of particular interest were insulators, regulatory elements first recognized in the early 1990s for their ability to act as local barriers to cis-acting elements (e.g., blocking the action of distal enhancers on promoters) (West, Gaszner, & Felsenfeld, 2002). By the turn of the century, CTCF had become a poster child as the first protein recognized as binding enhancer-blocking insulators in vertebrates (A. C. Bell, West, & Felsenfeld, 1999, 2001; Ohlsson, Renkawitz, & Lobanenkov, 2001; West et al., 2002). However, its mechanistic role in insulator activity remained a mystery (Ohlsson et al., 2001), making it a promising candidate for a possible role in TAD creation and modulation.
The first TAD papers in 2012 examined a host of factors as possible determinants of TAD formation, among which were CTCF binding sites found to be enriched at TAD boundaries (Dixon et al., 2012; Nora et al., 2012). However, noting that CTCF has many nonboundary bindings sites, the papers rationalized that the CTCF and cohesin proteins could not be primary determinants of TAD formation. Attempting to ascertain the importance of CTCF and cohesin loss on TADs, researchers performed partial depletion experiments in 2013 and 2014. CTCF depletion reduced insulation between adjacent TADs (Zuin et al., 2014) while cohesin depletion diminished long-range genomic interaction (Seitan et al., 2013; Zuin et al., 2014), relaxed chromatin domains (Sofueva et al., 2013), and dose-dependently affected cellular phenotype (Viny et al., 2015). Collectively, these results suggested that while CTCF and cohesin play important roles in nuclear architecture, they are not fundamentally necessary for TAD formation. However, subsequent ultra-high-resolution Hi-C analysis of the mammalian genome found that >86% of loop boundaries are enriched for CTCF and cohesin binding, implying a central role for both CTCF and cohesin in TAD formation (Rao et al., 2014). Critically, ChIP-seq, CRISPR genome editing, and Hi-C analyses established that the polarity of CTCF sites is a determinant of loop formation (de Wit et al., 2015; Guo et al., 2015; Rao et al., 2014; Sanborn et al., 2015; Tang et al., 2015; Vietri Rudan et al., 2015). CTCF's consensus DNA motif is not a palindrome; instead, it has polarity or directionality that is then also imparted to the CTCF protein. When considering a pair of CTCF sites, there are four possible conformations for their relative polarities: convergent (both sites oriented inward), divergent (both sites oriented outward), and tandem (both sites facing the same
留言 (0)