
記住我
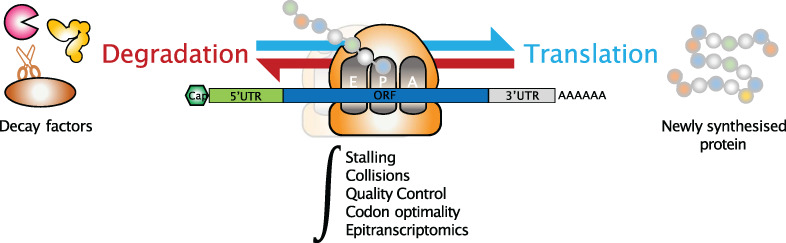
Messenger RNAs (mRNAs) are ephemeral intermediates between DNA, the carrier of genetic information, and proteins that display a functional role. Their primary sequence contains not only the information ribosomes need to synthesize proteins but also additional sequence and structural elements that regulate the translation process as well as other steps including transcription, splicing, polyadenylation, nuclear export, localization, and degradation. These elements can be located within untranslated regions (UTR) or in the coding region, owing to the degeneracy of the genetic code that allows some sequence flexibility. An intricate set of regulatory elements is therefore embedded within genes, under selective pressure, to play a specific role during the expression, function, or turnover of mRNAs.
mRNA translation is a sophisticated process that involves a large number of protein and RNA actors assisting ribosomal subunits in binding and assembling onto the mRNA. Thereafter, the ribosome translocates over the coding region and dissociates once the stop codon is reached, consequently releasing the newly synthesized protein. As such, translation is tightly regulated through a multitude of pathways involving cis-acting features and trans-acting factors that can affect the process in a transcript-specific or global manner. Although the initiation of translation is generally considered as the rate-limiting step, it has long been known that elongation is not a uniform process and termination is also tightly regulated, although these studies were technically challenging. The advent of high-throughput sequencing and the ribosome profiling protocol (Ingolia et al., 2009), together with structural biology approaches, have greatly contributed to the characterization of ribosome dynamics from the recruitment of the 40S to the elongation of 80S ribosomes across the open reading frame and the recycling of post-termination ribosomes. Collectively, these works have led to the discovery of new mechanisms involved in the regulation of translation and highlighted the numerous crosstalks between translation and other processes such as co-translational protein folding and mRNA turnover. This review summarizes recent findings that contribute to improve our understanding of the intricate relationship between mRNA decay and the translation process, with particular emphasis on how the translating ribosome has emerged as a central regulatory hub not only for the quality control pathways that degrade faulty mRNAs but also for canonical mRNA degradation.
2 TRANSLATION-DEPENDENT mRNA SURVEILLANCE PATHWAYSTo ensure high fidelity of gene expression, eukaryotic cells have evolved three major cytosolic mRNA surveillance pathways that rely on translation to recognize and trigger degradation of aberrant transcripts. These pathways, known as nonsense-mediated decay, non-stop decay, and no-go decay, respectively, signal RNAs with a premature termination codon (PTC), RNAs lacking a termination codon and RNAs containing diverse ribosome-stalling sequences (Powers et al., 2020; Shoemaker & Green, 2012). Despite their several mechanistic differences, the three mRNA surveillance pathways survey mRNA quality through careful inspection of the translation process, alerting when ribosomes are not progressing normally, in order to degrade aberrant mRNAs and their nascent polypeptides and to recycle ribosomes.
2.1 Nonsense-mediated decayThis surveillance pathway causes decay of transcripts harboring a PTC that can be brought in by genetic mutations or introduced into an mRNA through stochastic errors in transcription or splicing. In addition to its role in eliminating aberrant mRNAs with a PTC, nonsense-mediated decay (NMD) also degrades a large number of normal transcripts that contain an upstream open reading frame (uORF) or a long 3′-untranslated region (UTR; Kurosaki et al., 2019; Nasif et al., 2018). In all cases, it is assumed that RNA features downstream of a termination codon must interfere with translating ribosomes and, depending on the NMD trigger, the termination codon is defined as premature or genuine. The core NMD factors comprise a set of up-frameshift (UPF) proteins (UPF1, 2, and 3B) and suppressors with morphogenetic effects on genitalia (SMG) proteins (SMG1, 5, 6, and 7). The NMD machinery is thought to be recruited to target mRNAs because translation termination at a PTC is aberrant. In normal translation, ribosomes decode transcripts until they encounter a stop codon, usually present in the most 3′-exon in close proximity to the poly(A)-binding protein (PABPC1 in mammals, Pab1 in yeast) bound to the poly(A) tail. PABPC1 promotes translation termination by stimulating the recruitment of eukaryotic release factors eRF1 and eRF3a to the ribosomal A-site, which releases the nascent peptidic chain (Ivanov et al., 2016). Afterwards, ribosomes are dissociated into 40S and 60S ribosomal subunits that are recycled by the factor ABCE1 (Rli1 in yeast) with help of several initiation factors (Figure 1(a)).

Translation termination at a normal stop codon and at a premature termination codon. (a) Normal translation termination. Panel 1: The translating 80S ribosome, composed of 60S and 40S subunits, encounters the STOP codon (red STOP sign) located near the poly(a) tail bound by PABPC1 in mammals. Ribosomal A-site, P-site, and E-site are indicated. tRNAs are bound in the P-site and E-site. PABPC1 promotes the recruitment of the eRF1 and eRF3a release factors to the ribosomal A-site. Panel 2: After GTP hydrolysis by eRF3a, eRF1 triggers hydrolysis of the peptidyl-tRNA, releasing the completed nascent protein. This leaves a ribosome still bound to the mRNA, which has to be disassembled and recycled. Panel 3: The ribosome recycling factor ABCE1 is recruited and it supports, with help of other initiation factors, the dissociation of the 80S ribosome into 40S and 60S subunits, which can then be recycled for extra rounds of translation. (b) Translation termination at a premature termination codon (PTC) and induction of nonsense-mediated mRNA decay (NMD). Panel 1: The ribosome stops at a PTC located upstream of an EJC and distant from the normal stop codon. The presence of the NMD factor UPF3B at the EJC allows the association of eRF1 and eRF3a in the ribosomal A-site. Panel 2: A truncated protein product is released through the action of eRF3a and eRF1. Panel 3: Ribosome dissociation is mediated by ABCE1 together with UPF3B. Subsequently, UPF2 and UPF3B are thought to activate the SMG1 kinase for UPF1 phosphorylation. Panel 4: Phosphorylated UPF1 mainly associates with the SMG5-SMG7 complex which recruits decapping and deadenylation activities (1), followed by the exoribonucleases with 5′-to-3′ (XRN1) and 3′-to-5′ (exosome or DIS3L2) activities (2). In addition, phospho-UPF1 can recruit the endonuclease SMG6 that cleaves RNA in the vicinity of the PTC
In the case of a PTC, two predominant models of NMD activation have been put forward. The first, referred to as the exon junction complex (EJC) model, postulates that, during the pioneer round of translation, when a ribosome encounters a stop codon located sufficiently upstream of an EJC, a warning signal is sent to the NMD pathway to degrade the mRNA carrying a PTC (Nagy & Maquat, 1998; Thermann et al., 1998; Zhang et al., 1998). In this scenario, PABPC1 cannot interact with eRF3a due to their distance and, instead, the EJC allows the recruitment of NMD factors which, in turn, associate with eRF3a. Importantly, it has been reported that eRF3a interacts directly with UPF3B, but not UPF1 as previously thought, and UPF3B acts to dissociate post-termination ribosomes (Neu-Yilik et al., 2017). In addition, the ribosome recycling factor ABCE1, required for ribosome dissociation at a normal termination codon, is also essential for termination at a PTC, contrary to what was previously believed (Zhu et al., 2020). Indeed, loss of ABCE1 was found to induce an accumulation of unrecycled ribosomes in the 3′-UTR of NMD targets, leading to displacement of downstream EJC, followed by an impaired recruitment of the central NMD factor UPF1. Following proper ribosome dissociation, UPF1, activated by SMG1-mediated phosphorylation and aided by its partners UPF2 and UPF3B, can remodel the 3′-UTR and recruit the enzymes involved in mRNA decay (Figure 1(b)). The second model for NMD, called the faux UTR model, has been proposed in yeast. This model postulates that the inherently aberrant nature of premature translation termination allows the binding of UPF factors to mRNAs, thus triggering NMD. Supporting this model, early results showed that translation termination at a PTC is inefficient, likely because the PTC is distant from the poly(A)-binding protein Pab1 located at the 3′-end of the ORF (Amrani et al., 2004). In vivo experiments in yeast now substantiate these previous findings by demonstrating that the efficiency of translation termination increases as PTCs are closer to ORF 3′-ends, and furthermore this positional effect depends on Pab1 (W. Chan et al., 2020). The authors proposed from their results that NMD is activated as soon as the efficiency of termination is below a certain threshold.
By developing a method that allows real-time imaging of both translation and NMD of single mRNA molecules in live human cells, a recent work provided a unifying model for NMD activation by recapitulating most aspects of NMD, but this study also unveils that the mechanism of NMD activation is more complicated than previously believed (Hoek et al., 2019). This powerful method to study NMD kinetics and heterogeneity reveals that each ribosome (the first or one of the following ribosomes) that terminates translation at the PTC has an equal probability of triggering NMD. In addition, NMD occurs with the same probability during the pioneer round of translation (CBC-bound mRNAs) or during a later round (eIF4E-bound mRNAs). Surprisingly, for a given NMD substrate, a fraction ranging from 5 to 30% of mRNA molecules is resistant to NMD. This could be explained by some variability in splicing or EJC deposition. Contrariwise, NMD probability can vary substantially depending on several key parameters such as the distance between the PTC and the downstream EJC, the number of EJCs, or the mRNA sequence downstream of the PTC. On the other hand, changing the 3′-UTR length of the mRNA reporters did not have a major effect on NMD efficiency. This finding agrees with a recent study reporting that NMD in human cells occurs independently of stable ribosome stalling at PTCs (Karousis et al., 2020). To monitor ribosome density at stop codons, the authors developed a toeprinting assay based on in vitro translation of NMD reporter mRNAs with human cell lysates. Their results demonstrated a comparable ribosomal density at stop codons of NMD-sensitive and NMD-insensitive reporters. In line with this, ribosome profiling analyses also did not show any increase in ribosome occupancy at termination codon of endogenous NMD-sensitive transcripts, compared with NMD-insensitive mRNAs. Together, both studies do support that NMD is induced by a mechanism more complicated than just ribosome stalling at stop codons.
Another work was designed to define NMD targets and decay intermediates on a transcriptome-wide level (Kurosaki et al., 2018). Because NMD substrates are degraded by nucleases common to other RNA decay pathways and the UPF1 factor is involved in NMD but also in other decay mechanisms, the authors took advantage of the fact that, in human embryonic kidney (HEK) 293T cells, UPF1 is activated by phosphorylation exclusively during NMD and not during other decay pathways. The use of an antibody specific of phosphorylated UPF1 combined with high-throughput sequencing allowed them to selectively identify direct NMD targets and their decay intermediates. Importantly, their findings provided evidence that NMD initiates on mRNAs that are bound by one or more translating ribosomes. This strongly suggests that NMD does not occur in P-bodies (see Box ), in agreement with previous studies (Eulalio, Behm-Ansmant, Schweizer, & Izaurralde, 2007; Stalder & Mühlemann, 2009) but in disagreement with others (Durand et al., 2007; Sheth & Parker, 2006). Their data also support that phospho-UPF1 predominantly associates with the SMG5-SMG7 complex which recruits decapping and deadenylation activities, followed by the action of exoribonucleases with 5′-to-3′ (XRN1) and 3′-to-5′ (exosome or DIS3L2) activities. Alternatively, phospho-UPF1 can interact with the endonuclease SMG6, which cleaves in close proximity to the PTC (Figure 1(b)). Finally, the authors found that NMD decay intermediates were modified by uridylation at their 3′-ends by the poly(U) polymerases TUT4 and TUT7, which further increases 3′-to-5′ decay and also XRN1-mediated 5′-to-3′ decay.
2.2 No-go decayThe no-go decay (NGD) pathway acts to recognize and degrade transcripts containing sequence features that induce ribosome stalling during translation elongation (Doma & Parker, 2006; Tsuboi et al., 2012). The most prevalent stalling features into mRNAs comprise stable secondary structures as stem-loop motifs or GC-rich sequences, tracts of rare codons that leave the ribosomal A-site empty, or damaged RNA bases. Such obstacles that hinder the movement of ribosomes along mRNAs lead to ribosome stalling and potentially ribosome collision, which implies a collision between a stalled ribosome and the elongating ribosome behind it. Depending on the context, two different mechanisms evolved to cope with the event of ribosomal arrest. In the case of a stalled ribosome at the end of a truncated mRNA, which leaves empty the ribosomal A-site, it was found that the yeast complex Dom34-Hbs1 (or Pelota-Hbs1 in mammals) binds to the A-site and is thought to recruit an endonuclease for mRNA cleavage in the vicinity of the stalled ribosome (Doma & Parker, 2006). The ensuing cleavage products are degraded by either the Xrn1 exonuclease or the Ski-exosome complex. Some mechanistic details were obtained using high resolution ribosome profiling and biochemistry approaches in yeast (D'Orazio et al., 2019). The authors identified Cue2 as the primary endonuclease that cleaves mRNA precisely within the A-site of the stalled ribosome and they found that exonucleolytic decay of RNA intermediates proceeds primarily with Xrn1 rather than the exosome. Furthermore, the NGD process closely couples decay of faulty mRNAs with ribosome rescue and degradation of the nascent polypeptide. Dom34/Pelota and Hbs1, which are structural homologs of the canonical termination factors eRF1 and eRF3, recruit the ribosome-recycling factor Rli1/ABCE1 to promote dissociation of the stalled ribosome (Tsuboi et al., 2012). Because Dom34/Pelota is unable to manage peptidyl-tRNA hydrolysis, the nascent peptide remains attached to the 60S ribosomal subunit and is then handled by the ribosome-associated quality control (RQC) pathway for subsequent ubiquitination, extraction, and degradation by the proteasome (Shao et al., 2013; Verma et al., 2013; Figure 2(a)).

No-go decay and non-stop decay. (a) No-go decay (NGD). Panel 1: The ribosome stops before reaching the stop codon because it encounters a stalling feature such as a stem-loop motif, a GC-rich tract, or rare codons. These obstacles lead to ribosome stall and potentially ribosome collision. Two different mechanisms evolved to cope with this particular event of ribosomal arrest. The first mechanism, aimed to resolve a stalled ribosome, is illustrated on panels 2 and 3. The mammalian complex Pelota-Hbs1 (Dom34-Hbs1 in yeast) binds to the empty ribosomal A-site and recruits the ribosome recycling factor ABCE1 (Rli1 in yeast) and also the Cue2 endonuclease (in yeast), which cleaves the mRNA in the A-site of the stalled ribosome, further allowing exonucleolytic decay mediated primarily by the XRN1 exonuclease rather than by the exosome assisted with the Ski complex. Panel 3: NGD closely couples decay of faulty mRNA with ribosome recycling and degradation of the nascent polypeptide. Because Pelota-Hbs1 cannot hydrolyze the peptidyl-tRNA bound to the P-site, peptide release does not occur. The nascent peptide still attached to the 60S ribosomal subunit is handled by the ribosome-associated quality control (RQC) pathway and degraded via the proteasome. Panel 4 illustrates the second mechanism aimed to resolve collided ribosomes. This event induces conformational changes of the ribosome that are recognized by the ubiquitin ligase ZNF598 (in mammals). ZNF598 ubiquitinates several ribosomal proteins and these modifications are required for robust induction of NGD. (b) Non-stop decay (NSD). NSD relies on the same factors as NGD, but differs in its mRNA targets. Panel 1: Due to the lack of a stop codon, the ribosome translates a poly(A) sequence and translation elongation progressively slows down and eventually stops. Activation of NSD results from interactions between the positively charged lysine residues of the nascent polypeptide and the negatively charged exit channel of the ribosome. Panel 2: The complex Pelota-Hbs1 in the A-site recruits the endonuclease NONU-1 (in C. elegans, homolog of Cue2 in S. cerevisiae). The interactions between positively-charged residues with the exit channel result in the retention of the nascent peptide when the ribosome subunits are dissociated. Panel 3: Ribosome stalling caused by translation of poly(A) sequences is recognized by ZNF598. Ubiquitination of ribosomal proteins by ZNF598 are important modifications for ribosome rescue during NSD
A second major mechanism is employed during NGD to resolve ribosome collision events. This mechanism does not involve external factors interfacing with ribosomes, but it relies on direct modifications of ribosomes. When the trailing ribosome collides with the leading stalled ribosome, the complex formed by the two is called a disome. The structure of the disome was solved by cryo-electron microscopy (cryo-EM) and revealed a unique conformation suitable for recognition and modification of some ribosomal proteins by the ubiquitin ligase ZNF598 (Hel2 in yeast; Ikeuchi et al., 2019; Juszkiewicz et al., 2018). In particular, ubiquitination of the ribosomal protein uS10 (RPS20) was found to be required for elimination of the disome unit by the NGD pathway in yeast (Ikeuchi et al., 2019). Consistent with these findings, it was demonstrated that ribosome collisions are crucial for robust induction of the NGD process (Simms et al., 2017; Figure 2(a), Panel 4).
2.3 Non-stop decayDespite its name, the non-stop decay (NSD) pathway is not dedicated to the decay of all transcripts lacking a stop codon but primarily dedicated to the decay of transcripts inappropriately polyadenylated in their coding region, resulting in formation of nonstop mRNAs (Frischmeyer et al., 2002). Therefore, although they differ in their substrate specificity and ribosome stalling mechanism, NSD and NGD both degrade problematic RNAs with stalled ribosomes and collided ribosomes if severe ribosome slowdown. In line with this, NSD relies on the same factors as NGD to induce mRNA decay and ribosome rescue (Figure 2(b)). These include Dom34/Pelota-Hbs1 and Rli1/ABCE1 (Tsuboi et al., 2012). Recently, NONU-1 was identified in C. elegans as a novel endonuclease that induces RNA cleavage in the vicinity of stalled ribosomes during both NSD and NGD (Glover et al., 2020). Homology searches revealed that one homolog of NONU-1 in S. cerevisiae is Cue2, which was previously identified as the primary endonuclease functioning in NGD (D'Orazio et al., 2019). Mechanistically, earlier studies suggested that ribosome stalling during NSD was caused by the interaction of the nascent polypeptide, positively charged because of consecutive lysine residues encoded by the poly(A) sequence, with the ribosomal exit channel that is charged negatively (Koutmou et al., 2015; Lu & Deutsch, 2008). Further biochemical and structural studies in mammalian systems have elucidated the mechanism by which ribosomes stall during translation of a poly(A) tail (Chandrasekaran et al., 2019). By cryo-EM, the authors visualized a suboptimal peptidyl-tRNA conformation for peptide bond formation and a reconfiguration of the decoding center by the poly(A) mRNA that conflict with incoming aminoacyl-tRNA, thus impeding further elongation. Accordingly, they proposed that NSD is selectively activated upon coincident detection by the ribosome of poly-lysine chains in the exit tunnel and poly(A) tracts in the decoding center. Additionally, it was found that ribosome stalling caused by translation of poly(A) sequences is recognized by ZNF598. This E3 ligase, also involved in NGD, ubiquitinates the 40S ribosomal proteins eS10 (RPS10), uS10 (RPS20), and eS1 (RPS3A) in mammals (Figure 2(b), Panel 3), and these modifications are crucial to detect and resolve ribosomes that have stalled during decoding of poly(A) sequences (Garzia et al., 2017; Juszkiewicz & Hegde, 2017; Sundaramoorthy et al., 2017). In yeast, similar ubiquitinations catalyzed by Hel2 were also described, yet targeting distinct ribosomal proteins (Matsuo et al., 2017).
3 TRANSLATION INITIATION AND mRNA DECAYIn eukaryotic cells, coupling between mRNA translation and decay seems to be widespread and extends beyond the aforementioned mRNA surveillance mechanisms. While individual steps of translation and decay processes are rather well elucidated, less is known about the upstream determinants that control mRNA fate. Translation initiation is generally viewed as the most influential step in translation and numerous studies have attempted to address its impact on mRNA decay. Early observations in S. cerevisiae have shown that active translation initiation protects certain mRNAs from degradation (Schwartz & Parker, 1999). For those transcripts, inhibition of translation initiation by mutation of initiation factors led to higher rates of mRNA decay, associated with an increase in both decapping and deadenylation activities. A transcriptome-wide study in S. cerevisiae revealed that this relationship between mRNA translation initiation and decay is broadly generalizable (P. P. Chan & Lowe, 2016). By combining a minimally invasive metabolic labeling-based assay to measure mRNA decay rates with selective perturbations of the translation process, these authors found that slowing translation initiation caused a higher rate of transcript degradation on a transcriptome-wide level. Consistent with this global destabilization of mRNAs upon inhibition of translation initiation, they observed an enhanced formation of P-bodies, which are sites of mRNA storage and decay. By contrast, they found that slowing translation elongation led to an overall stabilization of transcripts and no change on P-body formation. Collectively, their results provide further evidence that translation initiation is an important determinant of mRNA stability in yeast. Two mutually non-exclusive models have been proposed to explain how translation may affect mRNA stability. The first model, referred to as the stalled ribosome-triggered decay model, predicts that a slow rate of translation elongation at suboptimal codons is perceived as a signal to trigger mRNA decay. The second model, referred to as the translation factor-protection model, predicts that the translation initiation machinery protects mRNAs from decay by a competition mechanism for mRNA binding. The data obtained by (P. P. Chan & Lowe, 2016) are consistent with mRNA decay being regulated by translation initiation but not elongation.
In higher eukaryotes, a study now provides a broad and comprehensive view regarding the impact of the 5′-UTR on mRNA translatability and stability (L. Jia et al., 2020). By designing a mRNA library of over one million 5′-UTR variants with a randomized sequence preceding a small uORF and downstream GFP, the authors identified several elements in the 5′-UTR that contribute to reporter mRNA outcomes in mammalian cells. While translation of the GFP-encoding main ORF preserves mRNAs from degradation, uORF translation was found to inhibit translation of downstream GFP, by triggering mRNA decay in a way reminiscent of NMD since this uORF-mediated decay depends on both UPF1 and translation. Additionally, enrichment of GGC motifs in the 5′-UTR inhibits translation and confers a shorter half-life to mRNAs. These sequence elements form RNA G-quadruplex (RG4) structures that impair the scanning process and relocate the RG4 reporter mRNAs into P-bodies for DCP2-dependent or XRN1-dependent decay. Finally, a 5′-UTR unstructured A-rich motif was uncovered as an internal ribosome-entry site to mediate cap-independent translation. This poly(A) tract element, while stabilizing mRNAs engaged in translation, destabilizes ribosome-free mRNAs likely by recruiting nucleases. Together, these findings reveal that the 5′-UTR contributes to a tight coupling between mRNA translation initiation and degradation. Furthermore, this coupling can occur through various mechanisms depending on the 5′-UTR sequence features.
4 THE RIBOSOME AS A HUB FOR mRNA DECAY, QUALITY CONTROL, AND STRESS SIGNALINGTo ensure protein homeostasis, eukaryotic cells have evolved co-translational quality control mechanisms that respond to abnormal translation by targeting the associated mRNA and nascent polypeptide chain for degradation and by recycling ribosomes (Joazeiro, 2019). Emerging evidence highlights crucial roles for the ribosome in quality control mechanisms, acting as a hub for recruiting factors that determine the fate of mRNAs and polypeptides. In addition, ubiquitin modifications at the ribosome emerge as part of the quality control pathways. Furthermore, recent work has revealed that the ribosome is a new central player to trigger various stress responses.
4.1 The ribosome and mRNA decayIt was long believed that mRNA degradation required the removal of ribosomes first until a pioneering study in yeast uncovered that mRNA decay could occur while transcripts were still bound by translating ribosomes (Hu et al., 2009). The prevailing model of cytoplasmic mRNA decay is that it starts with shortening of the poly(A) tail by the Ccr4-Not deadenylase complex and removal of the 5′-cap structure by the Dcp1–Dcp2 decapping complex. Then, mRNA decay proceeds through 5′-to-3′ degradation by the exoribonuclease Xrn1 and/or 3′-to-5′ exonucleolytic degradation by the exosome assisted with the Ski complex (Figure 3(a)). The study by Hu et al. showed that decapping and 5′-to-3′ exonucleolytic degradation take place on certain mRNAs still associated with ribosomes. Their findings indicate that removal of ribosomes is not a prerequisite for initiation of mRNA decay and, furthermore, suggest that Xrn1-mediated mRNA degradation from the 5′-end follows the last elongating ribosome. By using high-throughput sequencing of 5′-phosphorylated mRNA degradation intermediates, another work revealed that up to approximately 35% of transcripts in S. cerevisiae undergo co-translational decay, which is inferred from the characteristic 3-nucleotide periodicity pattern observed in the mRNA coding region (Pelechano et al., 2015). Additionally, the use of novel sequencing technologies to identify RNA decay intermediates has shown that co-translational mRNA degradation is much more widespread than previously thought (Ibrahim et al., 2018). Ibrahim et al. have developed a new transcriptome-wide method coined “Akron-seq” to simultaneously capture and sequence the 3′-ends of capped mRNAs (Akron3) or the 5′-ends of polyadenylated transcripts (Akron5) in human cells (Ibrahim et al., 2018). Their results showed that most 3′ and 5′ RNA ends are mapped within the mRNA body. Specifically, up to 63% of the 3′-ends of capped mRNAs are mapped within coding sequences and only 11% of capped transcripts contain a poly(A) sequence at their 3′-end, thus indicating that fragmented mRNAs are much more represented in cells than full-length RNA molecules. Importantly, relative positioning analyses of Akron reads with ribosome profiling data revealed that 5′-end and 3′-end reads exhibit a striking 3-nucleotide periodicity for all transcripts, suggesting that canonical mRNA degradation may be coupled with translating ribosomes. Furthermore, their positioning analyses stipulated that RNA intermediates are not generated by exonuclease activities but by a repeated ribosome-associated endonucleolytic activity, which cleaves translated RNAs at the exit site of mRNA ribosome channel. This cleavage generates a 5′-capped mRNA fragment with potentially active elongating ribosomes. When the leader ribosome on this 5′-capped fragment reaches the 3′-cleavage site, it undergoes a hard stall (the 3′-end of the mRNA being in the A-site) that induces a new endonucleolytic cleavage at the exit site of mRNA ribosome channel, generating a final degradation product of 16 nt in length. Ibrahim et al called “ribothrypsis” this novel process of ribosome-phased endonucleolysis, for which the endonuclease remains to be identified (Figure 3(b)). Beyond that, their study provides evidence that translation-dependent mRNA degradation is much more widespread than previously thought, as it is very widely used in canonical mRNA decay. Final ribothrypsis products are similar to the small ribosome footprints (15–18 nt) observed at known sites of mRNA truncation in yeast lacking the auxiliary exosome factor Ski2 (Guydosh & Green, 2014). However, final ribothrypsis products are naturally generated by a ribosome-associated endonuclease while the 5′-end of the small ribosome footprints observed in yeast are generated by the RNaseI treatment following cell lysis. It is, therefore, not known yet whether a mechanism similar to ribothrypsis is conserved in lower eukaryotes.

Conventional mRNA decay pathways in yeast and mammals. Cytoplasmic mRNA decay occurs independently of the translation process for the most part (a), but it can also proceed co-translationally (b). (a) The translation-independent mRNA decay starts with deadenylation by the CCR4–Not complex, followed by removal of the 5′-cap by the DCP1–DCP2 complex (Panel 1). Thereafter, mRNA decay proceeds through 5′-to-3′ degradation by the exoribonuclease XRN1 and/or 3′-to-5′ degradation by the exosome assisted with the Ski complex (Panel 2). (b) Co-translational mRNA decay is initiated when, for various reasons, a ribosome is not loaded by a new tRNA in its A-site. As above, mRNA decay starts with deadenylation by the CCR4–Not complex, then decapping by the DCP1–DCP2 complex, leading to exonucleolytic cleavage by XRN1 (5′-to-3′) and the ski-exosome complex (3′-to-5′). In mammals, the mRNA decay pathways are not redundant, but instead perform specialized functions. XRN1 predominantly mediates bulk mRNA decay with the aid of normal translation (Panel 1). The Ski-exosome complex functions in global mRNA surveillance and triggers mRNA decay in the case of aberrant translation events as stalled ribosomes at a premature stop codon (Panel 2). Alternatively, mRNA decay can occur through a repeated ribosome-associated endonucleolytic activity, for which the endonuclease remains to be identified (Panel 3)
Recently, Tuck et al. wanted to find out whether decay factors could interact with ribosomes in mammals, and to what extent mRNA decay was coupled to translation in higher eukaryotes (Tuck et al., 2020). Using mouse embryonic stem cells, they first performed a global comparison of transcripts bound by XRN1 or by the Ski complex helicase SKIV2L (homolog of yeast Ski2), and found a different specificity for a large number of transcripts, suggesting that the two cytoplasmic decay pathways could have specialized functions. Their results showed that XRN1 and SKIV2L are both recruited on translating ribosomes but their distribution is different, with XRN1 associating uniformly throughout the mRNA length and SKIV2L accumulating specifically on RNAs occupied with stalled ribosomes. This work, as a whole, sustains the idea that XRN1 mediates bulk mRNA decay with the aid of normal translation, whereas the Ski complex functions in global mRNA surveillance and assists the exosome in triggering mRNA decay in the case of aberrant translation events as stalled ribosomes (Figure 3(b)).
Consistent with mRNA decay occurring co-translationally, a cryo-EM structure of yeast Xrn1 bound to a ribosome was solved and the overall architecture of the Xrn1–ribosome complex is compatible with Xrn1 binding at the mRNA exit site of the ribosome (Tesina et al., 2019). Also, Schmidt et al. determined the structure of a yeast ribosome associated with the Ski2–Ski3–Ski8 complex and found that the Ski complex is positioned near the ribosomal mRNA entry tunnel (Schmidt et al., 2016). Additionally, a ribosome tagging method developed in mouse embryonic stem cells revealed that the mammalian “ribo-interactome” is composed of a repertoire of approximately 400 proteins including the expected ribosomal proteins and translation factors but also many unexpected proteins controlling diverse cellular processes (Simsek et al., 2017). Moreover, this dataset contains several RNA helicases including SKIV2L and UPF1, as well as certain subunits of the Ccr4-Not deadenylase complex and m6A-binding proteins, all of which are known to regulate the efficiency of mRNA translation and decay.
4.2 The ribosome and selective co-translational degradation of functional mRNAsBesides the role of ribosomes in orchestrating bulk mRNA degradation, other pathways involving co-translational degradation of selected functional transcripts exist. These involve ribosome interacting proteins that can either contact nascent peptides or specific RNA motifs within translated mRNAs to trigger their degradation.
Staufen-mediated mRNA decay (SMD) is a translation-dependent degradation pathway mediated by the mammalian double-stranded RNA (dsRNA) binding proteins Staufen1 and Staufen2 (Y. K. Kim et al., 2005; E. Park & Maquat, 2013). Staufen proteins bind dsRNA structures located in the 3′-UTR of specific mRNAs or resulting from intermolecular interactions between two different RNA species (de Lucas et al., 2014; Gong et al., 2013; Gong & Maquat, 2011; Ricci et al., 2014; Sugimoto et al., 2015). Staufen proteins interact directly with ribosomes as well as with UPF1, inducing phosphorylation of the latter and stimulating its helicase activity (Y. K. Kim et al., 2005; E. Park & Maquat, 2013). Similarly to NMD, SMD requires active translation of the target mRNA to trigger its decay. Interestingly, Staufen proteins and UPF2 were shown to bind to UPF1 at overlapping regions thus suggesting that their association to UPF1 is mutually exclusive (Gong et al., 2009). As a consequence, NMD and SMD were shown to be in competition, with the down-regulation of Staufen proteins activating NMD while UPF2 down-regulation stimulates SMD (Gong et al., 2009). However, biochemical reconstitution of UPF1 recruitment to Stau1 in vitro indicates a crucial role for UPF2 in facilitating UPF1 binding to Stau1 and its activation, indicating that the observed competition between NMD and SMD does not result from a competition of UPF2 and Stau1 to bind UPF1 (Gowravaram et al., 2019). Regarding its biological roles, SMD has been implicated in the regulation of myogenesis, adipogenesis, and in the local degradation of neuronal mRNAs upon their transport in axons, among others (Cho et al., 2012; Gong et al., 2009; J. Y. Kim et al., 2017).
Similarly to Staufen proteins, Regnase-1 is an RNA-binding protein with RNase activity that mediates translation-dependent mRNA degradation of specific transcripts through UPF1 (Mino et al., 2015). Regnase-1 binds to specific RNA stem-loops in the 3′-UTRs of inflammation-related mRNAs and plays an essential role in modulating the immune response (Cui et al., 2017; Kidoya et al., 2019; Mino et al., 2015; Tanaka et al., 2019; Uehata et al., 2013). Regnase-1 can bind to target mRNAs before they are translated but, in the absence of translation, this interaction does not result in mRNA degradation (Mino et al., 2019). Following the pioneer round of translation, UPF1 interacts with Regnase-1 and this association induces UPF1 phosphorylation and stimulates its RNA-helicase activity. As a consequence, UPF1 unwinds the RNA stem-loop structure bound to Regnase-1 and this conformational switch licenses Regnase-1 to cleave the target mRNA (Mino et al., 2019). Together, SMD-mediated and Regnase-1-mediated mRNA decay depend on specific RNA structures to recognize their target RNAs and exploit UPF1 to mediate translation-dependent mRNA decay, although through different mechanisms.
Translation-dependent mRNA degradation can occur independently of a sequence or a structural motif within the target mRNA but instead rely on the co-translational recognition of nascent peptides. Such a mechanism has been described for the degradation of mRNAs coding for secretory proteins whenever they fail to be correctly addressed to the endoplasmic reticulum (ER). Secretory proteins contain an N-terminal signal peptide that mediates co-translational recruitment of the signal recognition particle (SRP; Walter et al., 1981). Following its recruitment to the nascent peptide, the SRP induces a transient arrest of elongation and interacts with its receptor located in the rough ER to mediate the correct translocation of the nascent protein into the ER lumen before translation is resumed (Wolin & Walter, 1988, 1989). Mutations of the signal peptide that impair SRP recruitment lead to a decrease in protein expression and degradation of the corresponding mRNA (Karamyshev et al., 2014). Similarly, when SRP levels are down-regulated, the mRNAs coding for secretory proteins are destabilized within the cell. The absence of the SRP at the ribosome exit tunnel was shown to allow the Argonaute2 (Ago2) protein to interact with the nascent peptide and the ribosome to trigger mRNA degradation through a still uncharacterized mechanism that is independent of its slicing activity (Karamyshev et al., 2014). However, Ago2 is not required for the degradation of all mRNAs encoding secretory proteins that fail to recruit SRP, thus suggesting that other proteins could be involved in this degradation pathway (Pinarbasi et al., 2018). Furthermore, it is not clear how the cell discriminates between mRNAs coding for secretory proteins that fail to recruit an SRP from mRNAs coding for cytosolic proteins.
Another example of co-translational mRNA degradation through recognition of nascent peptides occurs for the regulation of tubulin expression. Indeed, the levels of alpha and beta tubulins are tightly controlled within cells, through an autoregulation mechanism that induces the degradation of tubulin mRNAs when the concentration of soluble tubulin proteins (i.e., non-polymerized) is above a certain threshold (Cleveland, 1988; Cleveland et al., 1981; Gasic et al., 2019). This regulation occurs co-translationally and depends on the recognition of the first four residues of the alpha and beta tubulin proteins (Met–Arg–Glu–Ile for β-tubulin and Met–Arg–Glu–Cys for α-tubulin) by the tetratricopeptide protein 5 (TTC5) (Bachurski & Cleveland, 1994; Gay et al., 1989; Lin et al., 2020; Pachter, 1987; Theodorakis & Cleveland, 1992; Yen et al., 1988). TTC5 associates with nascent tubulin peptides and contacts the ribosome near the exit tunnel through interactions with 28S rRNA and ribosomal protein uL24 (Z. Lin et al., 2020). Mutation of TTC5 residues that interact with ribosomes leads to loss of the regulation. The authors also found that, when soluble tubulin levels are low, the binding of TTC5 to nascent tubulin peptides is prevented by a still unknown factor. In contrast, the activity of this factor is inhibited whenever cells detect an excess of soluble tubulin, thus allowing TTC5 to bind to nascent tubulin peptides and ribosomes and to induce mRNA degradation. The precise mechanism by which TTC5 mediates mRNA degradation is yet to be characterized. It could rely on the recruitment of mRNA decay factors to TTC5-mediated stalled ribosomes or on the recognition of a specific ribosomal conformational change induced by TTC5.
Altogether, these studies highlight the crucial role of ribosomes in integrating multiple signals originating from the nascent peptide or from cis-acting elements within mRNAs to mediate co-translational degradation of functional mRNAs.
4.3 The ribosome and associated quality control mechanismsIn addition to the emerging role of the ribosome as a platform for a large number of interactors, ribosome ubiquitination appears to be crucial, particularly in the context of ribosome collisions. Obviously, collided ribosomes are detrimental to protein homeostasis and thus require mechanisms to cope with them. The vast majority of insights about ribosome collisions has been obtained from model substrates consisting of aberrant mRNAs with stalling sequences which, when translated, engender the leading ribosome to stall, causing collisions with trailing ribosomes (see the Sections 2.2 and 2.3 about the NGD and NSD mRNA surveillance pathways). A few studies have now sought to systematically identify the proteins associated with collided ribosomes. Widespread ribosomal collisions can be induced using low-doses of the elongation inhibitor emetine. Using quantitative proteomics on polysome fractions from cells treated with low-dose emetine, two groups identified the protein Endothelial differentiation-related factor 1 (EDF1) as being the new first-acting ribosome collision sensor (Juszkiewicz et al., 2020; Sinha et al., 2020). Cryo-EM structure reveals that EDF1 and its highly conserved yeast homolog Mbf1 bind at the disome interface close to the mRNA entry channel (Sinha et al., 2020). If collisions persist, EDF1 is needed for stabilization of both the E3 ubiquitin ligase ZNF598 and the protein Grb10-interacting GYF (glycine-tyrosine-phenylalanine) domain protein 2 (GIGYF2) at collided ribosomes (Juszkiewicz et al., 2020; Sinha et al., 2020). GIGYF2 and ZNF598 were previously found to interact with the eIF4E-homologous protein (4EHP) (Morita et al., 2012), which inhibits translation by competing with eIF4E for binding to the 5′-cap structure (Rom et al., 1998). Several findings suggest that both GIGYF2-4EHP and ZNF598 aim to minimize ribosome collisions: the translational repressor GIGYF2-4EHP blocks new rounds of translation onto problematic mRNAs and marks these transcripts for decay whereas ZNF598 catalyzes 40S ribosomal subunit ubiquitinations required for ribosome rescue by the ribosome-associated quality control (RQC) pathway (Hickey et al., 2020; Juszkiewicz et al., 2020; Sinha et al., 2020; Weber et al., 2020; Figure 4). In particular, ZNF598 is able to discriminate between transient or long-lasting collisions and is essential to resolve ribosome queuing that can form on mRNAs containing ribosome stalling sequences (Goldman et al., 2021). In addition to ZNF598, a second E3 ubiquitin ligase was reported to function in RQC. The makorin ring finger protein 1 (MKRN1) binds to prematurely polyadenylated transcripts and ubiquitinates the ribosomal protein eS10 and PABPC1 (Hildebrandt et al., 2019). While it is clear that ubiquitinations are key for proper rescue of stalled ribosomes, their precise role and temporal sequence are just beginning to emerge. It has been found that the ZNF598-catalyzed ubiquitinations are reversed by the deubiquitinating enzymes USP21 and OTUD3 (Garshott et al., 2020). Furthermore, the authors reported that ubiquitination events are hierarchically organized, suggesting the existence of an ubiquitin code on ribosomes. Alternatively, ubiquitination marks may serve as a scaffold for recruiting downstream quality control factors involved in resolution of ribosome stalls.

Ribosome-associated quality control mechanisms. Upon ribosome collision induced by problematic mRNAs, EDF1 is recruited first at the disome interface (Panel 1), and then it stabilizes the interaction of ZNF598, GIGYF2, and 4EHP with collided ribosomes (Panel 2). These proteins act in two distinct ways to minimize ribosome collision: (1) GIGYF2-4EHP represses initiation of new rounds of translation by competing with eIF4E for binding to the cap structure and marks problematic mRNAs for decay (Panel 3). (2) ZNF598 ubiquitinates several 40S ribosomal proteins and these ubiquitinations are required for the ribosome-associated qualit
留言 (0)