
記住我


Mitochondrial disorders are a heterogeneous group of disorders associated with a wide range of clinical phenotypes, which arise due to dysfunction of the mitochondrial respiratory chain. Mitochondrial diseases with childhood-onset are often severe and progressive, with substantial morbidity and mortality. However, there are rare forms that show remarkable spontaneous recovery. Two homoplasmic variants in tRNAGlu (m.14674T>C/G) have been shown to be associated with reversible infantile respiratory chain deficiency [1-3]. These children present with hypotonia, hyporeflexia, lactic acidosis, and severe muscle weakness from early infancy and often require assisted ventilation, features similar to those seen in the fatal infantile forms [4], but they recover spontaneously, usually within a few months. Muscle biopsy demonstrates complex IV (cytochrome c oxidase, COX)-deficient fibers and mitochondrial proliferation, and biochemically a combined complex I/IV deficiency is usually seen [1-6]. Electron microscopy findings include abnormal mitochondria, but a more detailed description of ultrastructural abnormalities is sparse. This entity has been given the name reversible infantile respiratory chain deficiency. The mechanism of the sudden onset and spontaneous recovery has not yet been determined, but the m.14674T>C/G is thought to impair mitochondrial translation. As most individuals who are homoplasmic for m.14674T>C/G do not show signs or symptoms of muscle disease at any stage, it has been suggested that additional heterozygous variants in nuclear genes interacting with mt-tRNAGlu may be a prerequisite for disease manifestation [7].
In this study, we present clinical and laboratory findings in seven individuals from four families with mitochondrial myopathy associated with the homoplasmic m.14674T>C variant. Longitudinal follow-up data indicate that myopathic features may persist into adolescence/adulthood and may even deteriorate in some cases, and that signs and symptoms of myopathy may develop insidiously during childhood. Furthermore, this study sought to further characterize the expression of the individual mitochondrial respiratory chain complexes at different stages of this disease, to widen the understanding of underlying pathological mechanisms.
2 MATERIALS AND METHODS 2.1 PatientsThis study included seven patients (P1–P7) from four families with mitochondrial myopathy associated with the homoplasmic m.14674T>C variant (Figure 1 and Table 1). Detailed clinical information for each patient is provided in the supplement file. All patients were Caucasians, but from different ethnic groups (Swedish and Iraqi). Four of the patients (P1-P3, P5) have been partly reported previously [1, 6-8].

Pedigrees of the four investigated families. Filled symbol represents individuals with mitochondrial myopathy associated with the homoplasmic m.14674T>C variant, dot represents carrier, # represents clinically examined individual. In Family 4, two disease-causing genetic variants (SYNE1, c.25448G>A and m.14674T>C) are present. %, amount of m.14674T>C; M, mutated SYNE1 sequence; P, patient; R, relative; WT, normal SYNE1 sequence
TABLE 1. Clinical findings Family Patient age at onset Infantile clinical presentation Longitudinal follow-up Family history 1P1/M
1 mo
1 mo: Vomiting, feeding difficulties, and failure to thrive
2 mo: Pneumonia and sepsis, muscle weakness and generalized hypotonia
Tube feeding because of swallowing difficulties. No ventilatory support
Increased urinary excretion of lactate, Blood lactate 4X unl
Serum CK increased
Exercise intolerance throughout childhood and adolescence
1.5 yr: Slightly delayed gross motor development, walked unsupported
2.5 yr : Motor function, muscle strength and cognitive development were normal
18 yr: Reduced exercise capacity (40% of normal). Exercise-induced hyperlactatemia. Motor function, muscle strength at rest and deep tendon reflexes normal. Ophthalmological and cardiological investigations normal. Serum CK slightly elevated
Sister with high-functioning autism
Mother (I:2, Figure 1) healthy, without signs or symptoms of myopathy or exercise intolerance
2P2/M
1 mo
1 mo: Increasing irritability and lethargy
2 mo: Generalized muscular hypotonia, feeding difficulties, and failure to thrive
Blood lactate 12X unl
Serum CK mildly increased
Mild liver involvement. A liver biopsy at 4 mo with mild steatosis but normal enzyme histochemical COX
Blood lactate has been normal after rise in neonatal period
2 yr : Normal mental and fine motor development. Delayed gross motor development
3 yr: Walked unsupported
5 yr: Improved with slight muscle weakness
14 yr: Decreased muscle strength with walking difficulties, unable to run. Serum CK 16X−25X unl
32 yr: Decline in ambulation. 6-min walk test with reduced capacity (65% of expected). Exercise intolerance with myalgia and muscle cramps. Serum CK 6X unl
Mother (I:2, Figure 1) without any symptoms of myopathyP3/M
1 mo
1 mo: Respiratory syncytical virus infection with feeding difficulties
Muscular hypotonia, swallowing difficulties, and respiratory failure. Transient CPAP treatment
Massive urine excretion of lactate (110X unl).
Blood lactate 4X unl
Serum CK normal
Gastrostomy feeding until 2 yr
1.5 yr: Slightly delayed gross motor development, walked unsupported. Delayed speech development because of dysarthria
12 yr: Residual muscle weakness with a positive Gower's sign, exercise intolerance, decline in ambulation. Mild ptosis, increased lumbar lordosis, weak tendon reflexes. Serum CK normal
Cardiological and ophthalmological investigations normal
Maternal nephew of P2
Mother (II:2, Figure 1) without any symptoms of myopathy
P4/F
2 mo
2 mo: Feeding difficulties, dysphagia, and failure to thrive. Tube feeding was required for nutrition. Muscular hypotonia
Blood lactate 2X unl
Serum CK normal
Slightly delayed gross motor and speech development
3.5 yr: Exercise intolerance, dysarthria, difficulties swallowing solid food. Motor function, muscle strength at rest and deep tendon reflexes were normal
Normal blood lactate, serum CK 1.2X unl
Maternal half-sister of P3
Mother (II:2, Figure 1) without any symptoms of myopathy
3P5/F
3 wk
3 wk: Failure to thrive with poor feeding, vomiting and lactic acidosis
3 mo: Profound muscular hypotonia and swallowing difficulties which required gastrostomy
19 mo: Slightly delayed early gross motor development, walked unsupported
8 yr: Normal mental and fine and gross motor development
Required persistent feeding by gastrostomy and had exercise intolerance into adolescence
15 yr: Reported asymptomatic
25 yr: Episode of rhabdomyolysis (serum myoglobin 2X unl, serum CK >4X unl)
27 yr: Motor function, muscle strength at rest, and deep tendon reflexes were normal. Increased serum CK level (>3X unl)
29 yr: No clinical examination was performed, the patient reported being asymptomatic
Mother (R1) has had no clinical signs or symptoms of myopathy, biopsy did not show any signs of mitochondrial myopathy or any other muscle disease 4P6/F
4 y
No signs or symptoms of myopathy in infancy4 yr: Presented with stumbling gait
6 yr: Poor balance, complained of leg pain
10 yr: Experienced muscle fatigue which deteriorated after periods of activity, feeding difficulties, thin stature
Her cognitive skills are assessed as being in the borderline range
P6 and P7 are two out of four children of consanguineous parents
Both parents and the two siblings are healthy and have no signs or symptoms of myopathy
The mother had three miscarriages
P7/F
Birth
Born at term with generalized hypotonia, absence of deep tendon reflexes. Flexion contractures of fingers, adducted thumbs, bilateral clubfeet, bilateral hip dysplasia. Required nasogastric tube feeding because of difficulties in sucking and swallowing
Blood lactate normal
3 mo: Deterioration with respiratory tract infection that required assisted ventilation, which later could be phased out to be used only during sleep
11 mo: Gastrostomy because of swallowing difficulties
Hypotonia, decreased movement, weak voice during her first year of life. Delayed fine and gross motor development
2 yr and 3 mo: Normal mental and fine motor development. Generalized hypotonia, more proximally than distally. Can sit but not stand. Soft muscles upon palpation. No deep tendon reflexes. Breathing support during sleep
Weight and length – 3 SD
Abbreviations: F, female; M, male; mo, month(s); P, patient; unl, upper normal levels; wk, week; yr, years.All protocols were approved by the Regional Ethical Review Board in Gothenburg.
2.2 Laboratory analysesMorphological and enzyme histochemical analyses of fresh frozen skeletal muscle biopsy specimens were performed according to established protocols [9]. For estimation of the proportion of COX-deficient fibers, double staining of COX and succinate dehydrogenase (SDH) was performed. Immunohistochemistry was performed as described previously [10] with antibodies against subunits of the five respiratory chain complexes (complex I: NDUFB8; complex II: SDHB; complex III: UQCRC2; complex IV: MTCO1; complex V: ATPB; as well as the mitochondrial marker VDAC1 (porin); Table S1). In P7, the expression of nesprin-1 was also investigated because of an identified SYNE1 gene variant (Table S1).
Biochemical analysis including spectrophotometric analysis was performed as previously described [11].
Western blot analysis was performed on proteins extracted from skeletal muscle tissue. Five μg of protein/well was separated on precast 4%–12% Bis-Tris gels (Thermofisher Scientific, Carlsbad, CA, USA) and transferred onto a polyvinylidene difluoride membrane. Antibodies targeted against subunits of the five respiratory chain complexes and VDAC1 were used. For P7, the expression of nesprin-1 was also investigated. For antibodies used, see Table S1. Proteins were detected using the Super Signal West Femto Substrate (Pierce, Rockford, IL, USA) and enhanced chemiluminescent detection (Fujifilm LAS-4000) was used for visualization. Coomassie staining of myosin heavy chain was used as a loading control.
Sanger sequencing was applied for mtDNA analysis in patients and relatives in families 1–3. Whole-genome sequencing was performed in four individuals of family 4 (P6, P7, and both parents). Confirmation analysis was performed by Sanger sequencing in the investigated individuals of family 4.
3 RESULTS 3.1 Muscle histopathology and biochemistryMuscle biopsy was performed in infancy in all patients except P6, who was 9 years (yr) of age at the time of biopsy. In three patients (P1, P2, P5), follow-up biopsies were also carried out. The results from enzyme histochemical, immunohistochemical, ultrastructural, and biochemical analyses are summarized in Table 2. At all occasions, all patients showed mitochondrial myopathy with a variable number of COX-deficient fibers. When analyzed by immunohistochemistry, there was a consistent deficiency of complex I subunit NDUFB8 and complex IV subunit MTCO1 (Figures 2 and 3; Figures S1–S11). The subunit SDHB of complex II was accumulated in accordance with mitochondrial proliferation, as was the mitochondrial membrane protein VDAC1 (porin). There was no deficiency of the subunit UQCRC2 of complex III or subunit ATPB of complex V in any investigated specimens. In addition to mitochondrial alterations, follow-up biopsies in P2 (age 14 yr) and P5 (age 8 yr) revealed unspecific myopathic changes including muscle fiber necrosis, regeneration, and endomysial fibrosis, consistent with a chronic myopathic degenerative process (Figures S5 and S9). Follow-up biopsies in P1 (age 18 yr), P2 (age 5 yr), and P6 (age 9 yr) showed mainly mitochondrial alterations with enzyme deficiency and mitochondrial proliferation (Figures 2B,C and 3B; Figures S2, S4, and S10). P7 at age 1 month (mo) showed marked variability of fiber size with markedly increased interstitial connective tissue in addition the frequent COX-deficient fibers and immunohistochemical deficiency of complex I and complex IV (Figures 3A and 7A–C; Figure S11). In P2, a type 2 fiber predominance was seen at 5 yr of age, whereas there was normal fiber type distribution nine years later at age 14. In contrast, the follow-up biopsy in P5 demonstrated solely type 1 fibers. The follow-up biopsy in P1 demonstrated normal fiber type distribution.
TABLE 2. Results from muscle biopsy Family Patient Age at onset Age at biopsy Muscle pathology and enzyme analysis Structural changes Ultrastructural mitochondrial changes COX− fiber (%) Immunohistochemical investigation Enzyme activities 1 P1/M 1 mo 4 moFiber size variability, lipid accumulation, RRFs
Figure S1
Large subsarcolemmal and intermyofibrillar collections of giant mito. with circular and tubular cristae, lipid accumulation
Figure 4A,B
60 ndCI: 1200
(ref. 4500–8600)
CIV: 1.9
(ref. 6.1–15)
18 yrMito proliferation, some RRF
Subsarcolemmal accumulation of partly giant mito. with irregular cristae and paracrystalline inclusions
Figure 4C,D
40NDUFB8 reduced (60%a)
MTCO1 reduced (partial)
SDHB increased
Figure 2C
nd 2 P2/M 1 mo 2 moFiber size variability, lipid accumulation, RRFs
Figure S3
Large intermyofibrillar accumulation of giant mito. with abnormal cristae, lipid accumulation
Figure 4E,F
95 ndCI: 0 (udl)
(ref. 4500–8600)
CIV: 0.9
(ref. 6.1–15)
5 yrSlight fiber size variability, some internalized nuclei, some fibers with mito. proliferation
Figure S4
Subsarcolemmal accumulation of elongated mito. with dense matrix
Figure 4G,H
5 ndCI: 4100
(ref. 4500–8600)
CIV: 9.1
(ref. 6.1–15)
14 yrMarked fiber size variability, internalized nuclei, fiber splitting, increased connective tissue, occasional muscle fiber necrosis, mito. proliferation
Figure S5
nd 60NDUFB8 reduced (80%a)
MTCO1 reduced (partial)
SDHB increased
CI: 3200
(ref. 4500–8600)
CIV: 7
(6.1–15)
P3/M 1 mo 1 moFiber size variability, lipid accumulation, RRFs
Figure S6
nd 80 ndCI: 1150
(ref. 4500–8600)
CIV: 0.54
(ref. 6.1–15)
P4/F 2 mo 4 moFiber size variability, lipid accumulation, RRFs
Figure S7
Large subsarcolemmal and intermyofibrillar collections of giant mito. with circular and tubular cristae, lipid accumulation
Figure 5A,B
75NDUFB8 reduced (80%a)
MTCO1 reduced (50%a)
SDHB increased
CI: 925
(ref. 4500–8600)
CIV: 1.1
(ref. 6.1–15)
3 P5/F 3 wk 1 moFiber size variability, lipid accumulation, RRFs,
Large subsarcolemmal and intermyofibrillar collections of giant mito. with abnormal cristae and enlarged, elongated mito. with dense matrix, lipid accumulation
Figure 5C,D
80NDUFB8 reduced (95%a)
MTCO1 reduced (60%a)
SDHB increased
Figure 2A
CI: 930
(ref. 4500–8600)
CIV: 0.22
(ref. 6.1–15)
8 yrFiber size variability, increased connective tissue, internalized nuclei, fiber necrosis and regeneration, mito. proliferation
nd 20NDUFB8 reduced (severe, regional)
MTCO1 reduced (partial)
SDHB increased
Figure 2B
CI: 6140
(ref. 4500–8600)
CIV: 14
(ref. 6.1–15)
4 P6/F 4 yr 9 yrFiber size variability, some fibers with lipid accumulation, mito. proliferation, some RRFs
Subsarcolemmal and intermyofibrillar collections of enlarged, partly elongated, mito. with single or multiple inclusions
Figure 5E,F
5NDUFB8 reduced (moderate, regional, 10%a)
MTCO1 reduced (mild, regional)
SDHB increased
Figure 3B
nd P7/F Birth 1 moPronounced fiber size variability, increased connective tissue, some fibers with internal nuclei, lipid accumulation, nesprin-1 absent from nuclear membranes
Intermyofibrillar collection of elongated mito. with dense matrix, lipid accumulation
Figure 5G,H
15NDUFB8 reduced (60%a)
MTCO1 reduced (10%a)
SDHB increased
Figure 3A
nd Note Enzyme activities: CI = nmol/min × mg protein; CIV = k/mg protein; k = rate constant. Abbreviations: CI, complex I; CIV, complex IV; COX−, COX-deficient fibers in COX/SDH staining; F, female; M, male; mo, month; mito., mitochondria; nd, no data; RRF, ragged-red fiber; udl, under detection limit; wk, weak; yr, year. a Proportion of fibers with complete lack of immunoreactivity.
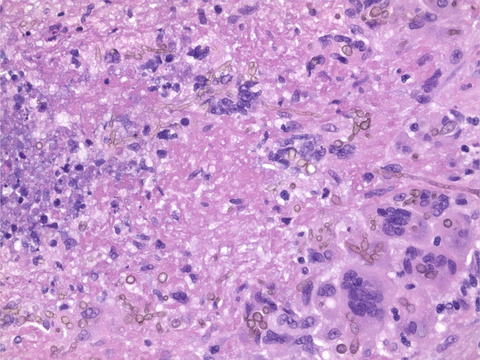
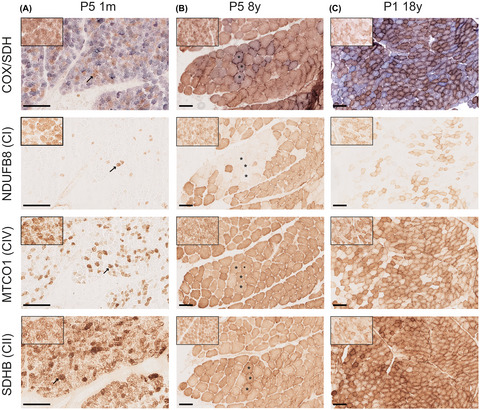
Serial sections of skeletal muscle of P5 and P1 at different ages. COX/SDH enzyme histochemistry with fiber lacking COX activity staining blue. Immunohistochemistry was used to visualize NDUFB8 (complex I), MTCO1 (complex IV), and SDHB (complex II). (A) P5 at 1 mo of age. A majority of the fiber show COX deficiency. A marked decrease of NDUFB8 and MTCO1 is seen, although the lack of MTCO1 is not as pronounced as the lack of NDUFB8. Some myofibers show high expression of SDHB. Identical fibers are marked with arrows. (B) P5 at 8 yr of age. A few fiber show partial COX deficiency. About 25% of the fiber lack NDUFB8 staining. These fibers show a regional distribution, rather than an even distribution. Some cells reveal a decrease in MTCO1 staining, but there is not a complete absence as seen at 1 mo of age. Occasional fibers show hyper-reactivity of SDHB. Identical fibers are marked with asterixis. (C) P1 at 18 yr of age. Numerous fibers show COX deficiency. A majority of the fibers lacks NDUFB8. Some fibers demonstrate decreased MTCO1 expression, only a few scattered fibers completely lack MTCO1. The fibers with abolished MTCO1 expression also completely lack NDUFB8. Some fibers show hyper-reactivity of SDHB. Insets show staining of a control aged 1.5 yr. Scale bars measure 100 μm

Serial sections of skeletal muscle of affected individuals in Family 4 (P7 and P6). COX/SDH enzyme histochemistry with fibers lacking COX activity staining blue. Immunohistochemistry was used to visualize NDUFB8 (complex I) and MTCO1 (complex IV). (A) P7 at 1 mo of age. Numerous fibers show COX deficiency. A majority of the fibers lack NDUFB8 staining and some fibers show reduced expression of MTCO1. Only a few scattered fibers completely lack MTCO1 expression. Insets show staining of an age-matched control. (B) P6 at 9 yr of age. Numerous fibers show COX deficiency. Numerous but not a majority of the fibers completely lack NDUFB8 expression with regional differences and some fibers show a decreased expression. Scattered fibers show a reduced of MTCO1 expression. Scale bars measure 100 μm
Biochemical analyses of the respiratory chain enzyme complexes revealed profound combined complex I and IV deficiency in all examined infant patients and milder defects at follow-up (Table 2).
3.2 Ultrastructural analysisElectron microscopic analysis of mitochondria in infant P1, 2, 4, and 5 biopsies demonstrated intermyofibrillar and subsarcolemmal accumulation of enlarged, frequently giant mitochondria with circular and tubular cristae in addition marked increase in fat (Figures 4A,B,E,F and 5A–D). In follow-up investigations of P1 at age 18 yr (Figure 4C,D), and P2 at age 5 yr (Figure 4G,H), and in P6 at age 9 yr (Figure 5E,F) the mitochondrial changes were seen as subsarcolemmal and partly intermyofibrillar accumulation of mitochondria with paracrystalline and other inclusions and frequently elongated mitochondria with dense matrix. P7 showed in addition to abnormal elongated mitochondria with dense matrix (Figure 5G,H), numerous atrophic fibers, myofibrillar disorganization and multilobulated nuclei.

Electron micrographs of skeletal muscle from P1 and P2. (A and B) P1 at age 4 mo showing increased fiber size variability and large fat droplets (arrowheads) in some fibers. Collections of giant mitochondria with circular and tubular cristae are present in many fibers (arrows). (C and D) P1 at age 18 yr showing subsarcolemmal collections of mitochondria with paracrystalline inclusions (arrows). (E and F) P2 at age 2 mo showing an increased variability in fiber size and large fat droplets (arrowheads) in several fibers. Collections of giant mitochondria with tubular and circular cristae are present in many fibers (arrows). There is also a cytoplasmic body present in one fiber (asterisk). (G and H) P2 at age 5 yr showing subsarcolemmal collections of mitochondria with elongated shape and dense matrix (arrow). There is also a core structure in a fiber (asterisk) and increased interstitial collagen (col)

Electron micrographs of skeletal muscle from P4, P5, P6 and P7. (A and B) P4 at age 4 mo showing collections of giant mitochondria with circular and tubular cristae (arrows). (C and D) P5 at age 1 mo showing muscle fibers with subsarcolemmal accumulation of glycogen and collections of large mitochondria (arrows). The mitochondria show circular and tubular cristae with dense matrix. (E and F) P6 at age 9 yr showing subsarcolemmal and intermyofibrillar collections of large mitochondria multiple inclusions (arrows). (G and H) P7 at age 1 mo showing increased fiber size variability and increased interstitial collagen (col). Some fibers show increased amounts of elongated intermyofibrillar mitochondria with dense matrix (arrows)
3.3 ImmunoblottingTo analyze possible effects of the m.14674T>C variant on protein expression of the individual respiratory chain complexes, protein expression in homogenates of skeletal muscle from all patient biopsies, from the mother of P5 (R1), and two healthy controls (C1 and C2, both one year of age) was determined (Figure 6). This revealed a severe reduction of NDUFB8 in the patients’ muscle biopsy specimens in infancy. Results from analysis of an additional subunit of complex I, NDUFS3, in family 4 showed a similar pattern as NDUFB8 (Figure S12). Expression of MTCO1 was also downregulated, but not to the same extent as NDUFB8. These results are in agreement with the results from immunohistochemical analyses.

Immunoblotting shows decreased levels of the complex I subunit NDUFB8 and the complex IV subunit MTCO1 in patients’ muscle biopsy specimens in infancy. In follow-up biopsies, when the patients showed clinical improvement (P2 5y and P5 8y), as well as in the healthy mother of P5 (R1), the expression levels of NDUFB8 and MTCO1 were normal. The expression of both NDUFB8 and MTCO1 was decreased in P2 at age 14 yr, while only NDUFB8 expression was decreased in P1 at 18 yr of age. Coomassie staining of the myosin heavy-chain band (MHc) serves as loading control. C, control, P, patient, R, relative
In follow-up biopsies, when the patients had recovered (P2 5 yr and P5 8 yr), as well as in the healthy mother of P5 (R1), the expression levels of NDUFB8 and MTCO1 were normal. However, the expressions of both NDUFB8 and MTCO1 were decreased in P2 at 14 yr of age, when his muscle function had deteriorated. This finding is in concordance with histology results showing an increase in COX-deficient fibers. In the follow-up biopsy of P1 at 18 yr of age, the expression of NDUFB8 was reduced, while the expression of MTCO1 was normal. Some of the patients showed an increase in the expression of SDHB (P2 at 2 mo of age, P3, P4, and both biopsies of P5 and P1), consistent with muscle biopsy findings of mitochondrial proliferation (Figure 6). These biopsies also showed an increase in VDAC1 expression, albeit not as immense as the increase in SDHB expression, compared to controls.
To determine whether the nonsense variant in SYNE1 affected protein translation, protein levels in skeletal muscle of P7 as well as two age-matched controls were examined using an antibody, MANNES1E, directed against both the nesprin-1 giant isoform as well as the short nesprin-1α2 isoform (Table S1). The protein expression of nesprin-1 in P7 was entirely abolished (Figure 7F). In agreement with this finding, immunohistochemistry with an antibody against nesprin-1 (MANNES1A) demonstrated absence of the normal distinct staining of the nuclear membrane (Figure 7D,E).

Muscle pathology and Nesprin expression in P7. (A–C) Quadriceps muscle at 1 mo of age. There is marked variability of fiber size (range: 5–40 µm) and marked increase in connective tissue (A, H&E; B, Gomori trichrome; C, NADH-tetrazolium reductase; Scale bars measure 40 µm). (D and E) Immunohistochemistry using MANNES1A that recognizes the C-terminal region of nesprin-1-giant as well as nesprin-1-α2, the short isoform of nesprin-1, shows distinct staining of the nuclear rim (arrowheads) in an adult biopsy without signs of muscle disease, serving as a control (D). No such staining could be detected in P7 (E), (scale bars measure 50 μm). (F) Western blotting using MANNES1E showing that the expression of nesprin-1 was completely abolished in P7. Coomassie staining of the myosin heavy-chain band (MHc) serves as loading control
3.4 Genetic analysisP1–P3 and P5 have previously been reported to carry the homoplasmic m.14674T>C variant in MT-TE [1, 6]. In P4, sequencing of all mitochondrial tRNA genes revealed the same alteration. The mothers of P1, P2, P3, P4, and P5 carried the homoplasmic m.14674T>C variant, but it was absent in the father of P5 (Figure 1).
Whole-genome sequencing was conducted in four family members of family 4, including P6, P7, and their parents, in reference to the clinical findings of myopathy and arthrogryposis and mitochondrial changes on muscle biopsy in P7 and the mitochondrial myopathy in P6. In P7, two pathogenic variants were found, one homozygous variant in SYNE1 (c.25448G>A, NM_033071.3) resulting in a premature stop codon p.Trp8483* not reported in the Genome Aggregation Database (gnomAD), as well as the previously reported m.14674T>C variant. The parents were heterozygous for the SYNE1 variant and both of them were homoplasmic for the m.14674T>C variant. P6 carried the m.14674T>C variant in tRNAGlu (Figure 1). No homozygous- or compound heterozygous-predicted pathogenic variants were detected in EARS2, TRMU, QRSL1, GOT2, GLS, or MSS51, genes known to interact with mt-tRNAGlu. Damaging variants in such genes have been suggested to be a prerequisite for development of reversible infantile respiratory chain deficiency in carriers of the homoplasmic m.14674T>C variant [7]. Further filtering for variants in nuclear genes interacting with mt-tRNAGlu predicted as deleterious identified one heterozygous variant in MTO1 (c.1933C>A, NM_012123.4; p.Arg645Ser) in P7 and her mother.
4 DISCUSSIONReversible infantile respiratory chain deficiency was first d
留言 (0)