
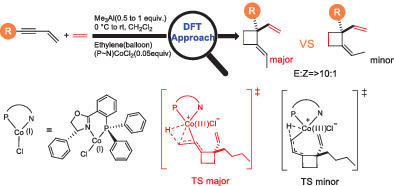
記住我


When different proposals exist (or can reasonably be formulated) for the size of the unit cell (in terms of number of atoms) and space group of crystalline compounds, a strategy for exploring with simulation methods the various cases and for investigating their relative stability must be defined. The optimization schemes of periodic quantum mechanical codes work in fact at fixed space group and number of atoms per unit cell, so that only the fractional coordinates of the atoms and the lattice parameters are optimized. A strategy is here presented, based on four standard tools, used synergistically and in sequence: (1) the optimization of inner coordinates and unit cell parameters; (2) the calculation of the vibrational frequencies not only at  , but also at a set of
, but also at a set of  points (in the example presented here they are eight, generated by a shrinking factor 2), looking for possible negative wavenumbers. The latter correspond to maxima, rather than minima, along the coordinate described by the corresponding normal mode; (3) the exploration of the total energy along the mode with negative wavenumber, looking for the minimum of the curve; (4) the identification of the new space group corresponding to the reduced symmetry resulting from the previous step. The strategy is illustrated with reference to the KMnF3 perovskite compound, for which many space groups are proposed in the literature, ranging from cubic
points (in the example presented here they are eight, generated by a shrinking factor 2), looking for possible negative wavenumbers. The latter correspond to maxima, rather than minima, along the coordinate described by the corresponding normal mode; (3) the exploration of the total energy along the mode with negative wavenumber, looking for the minimum of the curve; (4) the identification of the new space group corresponding to the reduced symmetry resulting from the previous step. The strategy is illustrated with reference to the KMnF3 perovskite compound, for which many space groups are proposed in the literature, ranging from cubic  to tetragonal
to tetragonal  and orthorhombic (Pnma and Cmcm) down to monoclinic (P21/m). The corresponding primitive cells contain 5, 10, and 20 atoms in the various cases, and the point symmetry reduces from 48 to 4 operators. In nature, the KMnF3 phase transitions also include the magnetic phases. For simplicity, here we limit the analysis to the ones that take place between ferromagnetic phases, as they are sufficiently rich for illustrating the proposed strategy. As the total energy differences involved can be as small as, say, 10–50 μHartree, a high numerical accuracy at each one of the steps mentioned above is required. The present calculations, performed with the CRYSTAL code, by using an all electron basis set and the Hartree-Fock and B3LYP functionals, document such an accuracy. The energy difference between the tetragonal
and orthorhombic (Pnma and Cmcm) down to monoclinic (P21/m). The corresponding primitive cells contain 5, 10, and 20 atoms in the various cases, and the point symmetry reduces from 48 to 4 operators. In nature, the KMnF3 phase transitions also include the magnetic phases. For simplicity, here we limit the analysis to the ones that take place between ferromagnetic phases, as they are sufficiently rich for illustrating the proposed strategy. As the total energy differences involved can be as small as, say, 10–50 μHartree, a high numerical accuracy at each one of the steps mentioned above is required. The present calculations, performed with the CRYSTAL code, by using an all electron basis set and the Hartree-Fock and B3LYP functionals, document such an accuracy. The energy difference between the tetragonal  and cubic
and cubic  phases is 225 μHartree, with a volume reduction of 0.58 Å3; the differences between the orthorhombic and tetragonal phases are an order of magnitude smaller, being 23 μHartree and 0.06 Å3 for total energy and cell volume, respectively.
phases is 225 μHartree, with a volume reduction of 0.58 Å3; the differences between the orthorhombic and tetragonal phases are an order of magnitude smaller, being 23 μHartree and 0.06 Å3 for total energy and cell volume, respectively.
留言 (0)