
記住我





In our previous study, we reported that short-term treatment with the autophagy-activating drug rapamycin negatively impacts virion release for nef-deficient HIV-1 NL4-3 in many cell types, including HEK293T, THP-1-derived macrophages, primary CD4+ T cells and Jurkat CD4+ T cells [28]. To evaluate the effect that autophagy poses on HIV fitness over several rounds of replication, we infected Jurkat cells with either HIV-1 NL4-3 or NL4-3 Δnef and maintained a constant concentration of rapamycin (6.5 μM) for 72 h. Virus replication was monitored every 24 h by measuring the levels of the capsid protein p24 (CA) released to the supernatant by p24 antigen-capture ELISA. Whereas rapamycin caused a 5-fold reduction in the replication of wild type NL4-3 72 h post-infection, this effect was magnified for the nef-defective virus, resulting in a 60-fold defect in its replication kinetics (Fig. 1A). Western blot analyses of the cell lysates showed that rapamycin treatment promoted the conversion of LC3-I into LC3-II (which serves as a measure for autophagy flux), confirming that autophagy was effectively activated. Besides this effect on LC3, rapamycin caused a concomitant defect in the emergence of Gag (p55) (Fig. 1B). However, in line with our previous findings [28], the presence of Nef counteracted both reduction of Gag levels and autophagy flux. This last effect is evidenced by the fact that, even in the presence of rapamycin, cells infected with wild type NL4-3 exhibited an accumulation of LC3-I, whereas in the Δnef-infected cells autophagy flux proceeded normally (Fig. 1B; see quantifications). To verify that rapamycin-induced autophagy is responsible for the defect in Gag levels and, in consequence, in virion production, similar assays were performed in the presence of 3-methyladenine (3-MA), a drug that blocks autophagy initiation [37, 38]. Since this compound can also trigger autophagy when used during prolonged treatments [39], 3-MA and rapamycin were added to the cultures for only 6 h. In this case, virion production was expressed as the percentage of maximal release relative to the DMSO-treated samples. Consistent with our previous work [28], addition of 3-MA prevented the activation of autophagy mediated by rapamycin, consequently rescuing virion production (Fig. 1C, D). To further evaluate the physiological relevance of these observations, we performed parallel assays in primary CD4+ T cells obtained from three healthy donors. Similar to the results obtained in Jurkat cells, rapamycin treatment successfully restricted HIV replication. Once again, the impact on virus replication was associated with an increase in autophagy flux and a defect in the emergence of Gag over time. Although the degree of restriction was not as striking as in the Jurkat system, autophagy activation posed a bigger hurdle for HIV Δnef than for wild type HIV (Fig. 1E, F). Likewise, treatment with 3-MA prevented autophagy activation and rescued Gag and virion levels (Fig. 1G, H). Hence, these findings indicate that the pharmacological activation of autophagy limits HIV replication in T cell lines and primary CD4+ T cells, and further confirm that Nef is an autophagy antagonist.
Fig. 1
Autophagy restricts HIV replication in Jurkat and primary CD4+ T cells. A, B Jurkat cells and E, F primary CD4+ T cells were infected with HIV-1 NL4-3 or NL4-3 Δnef and treated with rapamycin (6.5 μM) for 3 days. Supernatants were collected at each time point and were analyzed by p24 antigen-capture ELISA to determine relative viral replication. Jurkat cells (C, D) and primary CD4+ T cells (G, H) were infected with HIV-1 NL4-3 or NL4-3 Δnef and treated with DMSO, rapamycin (6.5 μM) or rapamycin and 3-MA (3 mM) for 6 h. Next, supernatants were collected and analyzed by p24 antigen-capture ELISA to determine relative viral replication. Data represents the mean and SEM of three independent replicates and significantly different values are indicated by asterisks (*P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001). Cell lysates from Jurkat cells (B, D) and primary CD4+ T cells (F, H) were analyzed by western blotting for Gag/p55, LC3-I/LC3-II and ACTB (β-actin) for each time point. Blots are representative of three independent experiments
Autophagy specifically targets HIV Gag for autolysosomal degradationOur results indicate that autophagy represents an important barrier for HIV replication. Therefore, we sought to determine what specific event in the virus life cycle autophagy is intersecting. The data presented in Fig. 1 indicate that autophagy activation is associated with a reduction in Gag (p55). Since Gag plays a crucial role in the recruitment of components for virion assembly [40,41,42], we hypothesized that autophagy causes defects in particle biogenesis by targeting Gag for elimination. Because autophagy maturation involves the fusion between autophagosomes and lysosomes, where the acidic pH and the presence of specialized proteases cause cargo degradation, we first assessed whether the pharmacological inhibition of lysosomal function could prevent the rapamycin-associated depletion of Gag. For this, we used HEK293T cells, since our previous work demonstrated that the restrictive role of autophagy on Gag levels and virion production—as well as the counteracting effect of Nef—is observed regardless of the cell type investigated, even in primary cells [28]. In addition, HEK293T cells are easier to manipulate, which is an advantage for mechanistic studies. Cells were transfected with the HIV-1 NL4-3 ∆nef proviral DNA, since this clone is more susceptible to autophagy restriction. 24 h later, the medium was replaced, and cells were treated with rapamycin for 12 h in the presence and absence of chloroquine (lysosomal inhibitor [43]). To rule out any potential degradation of Gag through the proteasomal pathway, we also included cells treated with rapamycin and ALLN, a proteasomal inhibitor [44]. As previously observed, Gag levels were reduced in cells treated with rapamycin (Fig. 2A; lane 1 vs. 2). The addition of chloroquine effectively blocked autophagy maturation, reflected by a significant accumulation of LC3-II. LC3-II coats the internal and external membrane of autophagosomes. Hence, upon fusion with lysosomes, LC3-II molecules on the internal membrane, as well as LC3-associated autophagy receptors (i.e., SQSTM1), are susceptible to degradation. However, impairment of lysosomal function prevents this process, consequently increasing the overall levels of LC3-II and SQSTM1. Under these experimental conditions, not only was LC3-II and SQSTM1 degradation prevented but also the rapamycin-induced degradation of Gag (Fig. 2A; lanes 3 and 4). By contrast, treatment with ALLN, had no impact on the rapamycin-dependent degradation of Gag or SQSTM1 (Fig. 2A; lanes 5 and 6). Therefore, these results confirm that the reduction of Gag caused by rapamycin is due to increased autophagolysosomal activity. Nevertheless, since HIV Gag is associated to cellular membranes through its myristoylated group in the N-terminus, it is plausible that the presence of Gag in autophagosomes may be coincidental, as a consequence of its membrane distribution. To rule this out, we assessed whether rapamycin-induced autophagy had a similar effect on the levels of gp120, another HIV protein that associates to cellular membranes. Remarkably, autophagy did not promote the degradation of gp120 (Fig. 2A), suggesting that autophagy targets Gag for degradation in a specific manner.
Fig. 2
Autophagy targets HIV Gag for degradation. A HEK293T cells were transfected with HIV-1 NL4-3 Δnef proviral DNA and treated with rapamycin (4 μM), chloroquine (60 μM) and/or ALLN (25 μM) for 12 h. 48 h later, lysates were analyzed by western blot for gp120, SQSTM1, p55, ACTB, and LC3. Densitometric analyses were performed to determine the relative ratios of gp120, SQSTM1 and p55. B HEK293T cells were co-transfected with Gag-EGFP, EGFP-LC3B or an empty vector. 48 h later, cells were harvested, and Gag was immunoprecipitated. The pulldown fraction was examined for SQSTM1 and LC3. Lysates were also analyzed by western blot for SQSTM1, Gag, LC3 and ACTB. C HEK293T cells were transfected with the HIV-1 NL4-3 provirus or an empty retroviral vector. 48 h later, cells were harvested, and LC3 was immunoprecipitated. The pulldown fraction was examined for LC3, SQSTM1, p55, gp120 and Nef. Lysates were also analyzed by western blot for gp120, SQSTM1, p55, Nef, LC3 and ACTB. D HEK293T cells treated with an irrelevant siRNA (si-ctr) or SQSTM1-specific siRNAs were transfected with HIV-1 NL4-3 Δnef proviral DNA. 48 h post-transfection, cells were harvested and endogenous LC3 was immunoprecipitated. The pulldown fraction was examined for LC3 and p55. Lysates were analyzed by western blot for SQSTM1, p55, LC3 and ACTB. E HEK293T cells were co-transfected with EGFP-LC3B and HIV-1 NL4-3 Δnef proviral DNA. Cells were exposed for 4 h to rapamycin (4 μM) or DMSO prior to microscopy visualization for EGFP-LC3 (green), Gag (red) and the nuclei (blue). Scale bar: 10 μm. All images are representative of three independent experiments
Next, the role of autophagy in targeting Gag for elimination was investigated by co-immunoprecipitation (co-IP) as well as fluorescence microscopy studies. We reasoned that if Gag is redistributed to autophagosomes by means of an autophagy receptor, we should detect a physical interaction—even if it is indirect—between LC3 and HIV Gag. For this, HEK293T cells were co-transfected with the fusion proteins Gag-EGFP and EGFP-LC3. 48 h later, cells were harvested, and lysates were subjected to immunoprecipitation. In this case Gag-EGFP was pulled down with a Gag-specific antibody, and its association with EGFP-LC3 and SQSTM1 was then analyzed (Fig. 2B). Of note, although the Gag and LC3 constructs both contain EGFP, IPs and blots were performed with antibodies against Gag and LC3 and not against EGFP. In order to evaluate if the proteins present in the pulldown fraction were the result of unspecific binding with the magnetic beads employed in these IPs, a control consisting of the cell lysates plus the magnetic beads, but no antibody, was included (beads ctr). Also, to discriminate between the heavy and light chains of the antibody used in the IP and the proteins of interest, an IgG control consisting of lysis buffer and beads coated with the antibody (IgG ctr) was included. Our data revealed that Gag-EGFP interacts with the autophagosome-associated proteins EGFP-LC3 and SQSTM1 (Fig. 2B). This indicates that Gag is recruited to LC3-coated autophagosomes, possibly by means of the adaptor protein SQSTM1. However, because in these assays we used Gag and LC3 constructs fused with EGFP, and EGFP is well known to dimerize [45, 46], we performed a similar experiment, with a more physiological approach, to verify that association between Gag and LC3 is not an artifact due to EGFP oligomerization. For this, endogenous LC3 was immunoprecipitated from HEK293T cells transfected with the full length wild-type NL4-3 provirus or an empty retroviral vector control. Unlike for Fig. 2A, we used the nef-competent NL4-3 clone to further examine whether viral proteins that inherently associate with cellular membranes, particularly by means of myristoylation (i.e., Gag, Nef), localize in autophagosomes coincidentally. In this case, the pulldown fraction was analyzed for the presence of SQSTM1, Gag, gp120 and Nef. As anticipated, the pool of LC3-interacting partners was positive for both SQSTM1 and Gag (Fig. 2C). However, no interactions between LC3 and Nef or gp120 were detected, which supports the notion that the autophagy-mediated recruitment of Gag is specific (Fig. 2C). Finally, to determine whether SQSTM1 plays a role in the elimination of Gag, similar assays were performed in cells depleted of this autophagy receptor. For this, HEK293T cells were transfected with a control siRNA or siRNAs specific for SQSTM1 1 day prior to the transfection with the NL4-3 ∆nef provirus. 72 h later, cells were harvested, lysates were immunoprecipitated for LC3 and its association with Gag analyzed. Of note, the heavy chain of the antibody used in the IPs was detected in the IgG control sample, but it exhibits a different migration pattern than that of Gag (Fig. 2D, left lane). Whereas similar amounts of Gag were found in the pulldown fraction of both control and SQSTM1-knocked down cells, the amount of LC3-I present in the immunoprecipates of cells depleted of SQSTM1 was remarkably high. Hence, binding between Gag and LC3-I/II relative to the control was reduced by 50% in this cellular context. In addition, due to the lower SQSTM1 levels, and thus slower autophagy flux, the overall amount of Gag/p55 and CA/p24 in the whole cell lysates was higher than in the control cells (Fig. 2D). Yet, the amount of Gag that was immunoprecipitated was not proportionally enriched, which further supports the notion that the recruitment and elimination of Gag through autophagy is greatly influenced by SQSTM1. Remarkably, the fact that LC3-I is the most abundant variant pulled down in the IPs (Fig. 2C, D) indicates that Gag might interact with nonlipidated LC3.
The recruitment of Gag to autophagosomes was demonstrated by fluorescence microscopy analyses. For this, HEK293T stably expressing EGFP-LC3 were transfected with NL4-3 ∆nef proviral DNA. Gag was visualized using an Alexa-568 conjugated (red) secondary antibody and the nuclei was stained with DAPI (blue). In the absence of rapamycin, EGFP-LC3 displays a cytosolic distribution. However, upon autophagy activation, it becomes incorporated into nascent autophagosomes and is subsequently detected as green puncta [10]. Consistent with the notion that Gag might interact with LC3-I, Gag was found scattered throughout the cytoplasm following a similar pattern as the cytosolic EGFP-LC3. However, after rapamycin treatment Gag exhibited a punctuate localization highly overlapping with LC3-coated autophagosomes (Fig. 2E). Similar observations were obtained in Jurkat cells (not shown). Hence, these findings confirm that upon autophagy activation HIV Gag is targeted to autophagosomes.
Ubiquitination and myristoylation are required to target Gag for autophagy-mediated degradationIn order to identify the genetic determinants that facilitate the autophagic recruitment and degradation of Gag, we performed immunoprecipitation assays and assessed the steady-state levels of three mutants of Gag. First, we used a G2A-Gag mutant, which cannot become myristoylated and thus, loses its ability to bind to membranes [47,48,49,50,51]. Second, since autophagy cargo is often poly-ubiquitinated, we introduced alanine substitutions at lysine residues in Gag predicted to become ubiquitinated (K113A, K114A, K335A, K359A, and K418A) generating the Ub-Gag mutant [44]. A third mutant (G2A/Ub-Gag) that lacks both the ability to become myristoylated and ubiquitinated was also generated. For this assay, HEK293T cells were transfected with the wild-type Gag-EGFP or the Gag-EGFP mutants and their interaction with the endogenous autophagy machinery was assessed by immunoprecipitation. Compared to wild-type Gag, LC3 interaction with the single mutants was significantly reduced, and almost completely abrogated for the double Gag mutant (Fig. 3A)—the relative interaction with LC3 was measured by densitometric analyses (Fig. 3A; bottom graph). Moreover, unlike wild type Gag, no significant fluctuations in the steady-state levels of the Gag mutants, particularly the double mutant, were observed after treatment with rapamycin for 12 h (Fig. 3B). Therefore, these findings indicate that not only Gag is specifically targeted by the autophagy machinery for autolysosomal clearance, but also that Gag ubiquitination and association with membranes are crucial for its autophagy-mediated recognition and degradation.
Fig. 3
Gag ubiquitination and membrane association are required to target Gag for autophagy elimination. A HEK293T cells were transfected with Gag, G2A-Gag, Ub-Gag or G2A/Ub-Gag. 48 h later, cells were harvested and LC3 immunoprecipitated. The pulldown fraction was examined for LC3, SQSTM1 and Gag. Lysates were also analyzed by western blot for the levels of SQSTM1, Gag, LC3 and ACTB. Bottom graph: Densitometric analyses to determine the relative LC3-Gag interaction. Data represent the mean and SEM of 3 independent replicates. B HEK293T cells were transfected with Gag, G2A-Gag, Ub-Gag or G2A/Ub-Gag and treated with rapamycin (4 μM) for 12 h. The cell lysates were analyzed by western blot for Gag, ACTB, and LC3. All images are representative of 3 independent experiments. Significantly different values are indicated by asterisks ***P ≤ 0.001
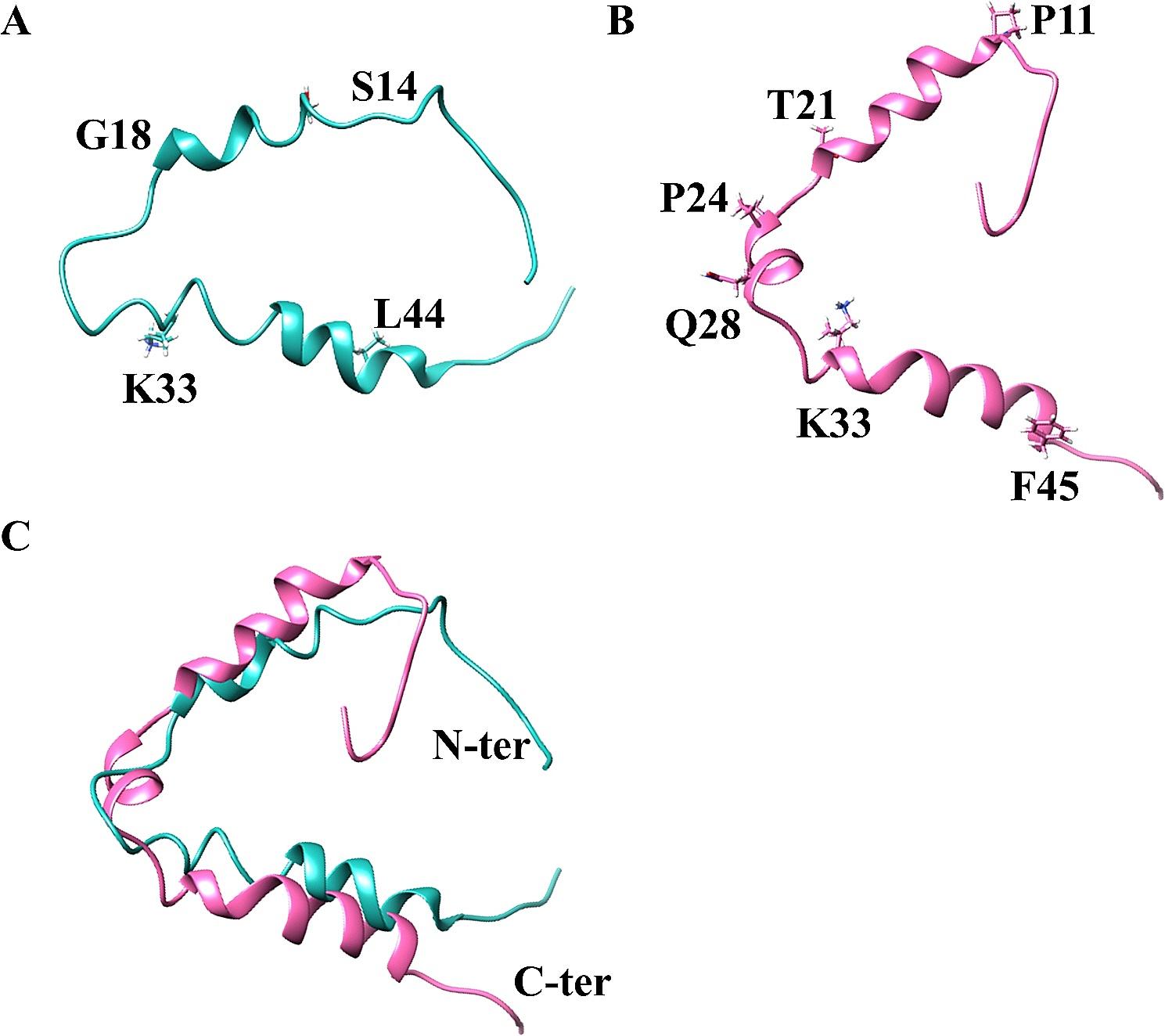
Residues comprising positions 40 to 57 in the N-terminal domain of HIV-1 NL4-3 Nef are required to block the early stages of autophagyUnlike NL4-3 Nef, our previous work revealed that SIVmac239 Nef cannot counteract autophagy restriction [28]. We took advantage of this fact to generate chimeric Nef proteins where we swapped individual functional domains between these two proteins with the goal of performing a loss-of-function assay and reveal the specific residues within NL4-3 Nef responsible for autophagy antagonism. For this, we replaced the N-terminus, globular core, flexible loop and C-terminus in NL4-3 Nef by the ‘inactive’ domains from SIVmac239 Nef, generating the chimeras I, II, III and IV (Fig. 4A). In order to determine whether these chimeras were able to intersect with autophagy, we first evaluated the impact of the resulting proteins on autophagosome biogenesis by flow cytometry assays, employing the same EGFP-LC3 construct used in Fig. 2. Besides the chimeras, we included SIVmac239 Nef and NL4-3 Nef as negative and positive controls, respectively. The principle of these assays relies on the fact that EGFP-LC3 binds to autophagosomes during their elongation, making EGFP-LC3 resistant to saponin treatments. Hence, after washing cells with a saponin-based wash buffer, the EGFP signal detected correlates with autophagosome formation [28]. 48 h post-transfection, cells were treated with rapamycin for 4 h prior to flow cytometry processing. With the exception of chimera I-transfected cells, which had similar levels of autophagosome formation as those expressing SIVmac239 Nef, cells transfected with chimeras II, III and IV displayed low autophagosome biogenesis (Fig. 4B and Additional file 1: Fig. S1). To corroborate these observations, we next analyzed the effect of these chimeras on the relative levels of LC3 lipidation by western blot. All Nef constructs, including these chimeras, were cloned into the expression vector pCGCG, which harbors EGFP from an internal ribosomal entry site [52, 53]. This feature was especially useful for these assays, since in some instances the mutagenesis of the native proteins modified the epitope sequence where the anti-HIV/SIV Nef antibodies bind. Therefore, we analyzed transfection efficiency and the expression of our constructs by monitoring the levels of EGFP, as in previous studies [28, 52, 54,55,56,57,58]. Of note, expression of EGFP from this construct is cytosolic and, thus, does not interfere with our quantification of EGFP-LC3-containing autophagosomes, since it is washed away upon saponin treatment [28]. Consistent with the flow data, NL4-3 Nef, together with the chimeras II, III, and IV, reduced LC3 lipidation even upon stimulation with increasing concentrations of rapamycin (Fig. 4C). However, chimera I, as well as the negative control SIVmac239 Nef, showed significantly higher ratios of LC3-II:I and, thus, normal autophagy flux (Fig. 4C; see quantifications underneath the blots), which indicates that the capacity of NL4-3 Nef to block LC3 lipidation and formation of autophagosomes resides somewhere along the N-terminal domain of the protein.
Fig. 4
Residues 40–57 in the N-terminal domain of NL4-3 Nef are required to counteract autophagy initiation. A, D Schematic representation of the domains replaced for the generation HIV-1 NL4-3 and SIVmac239 Nef chimeras. B, E HEK293T cells were co-transfected with EGFP-LC3B and the different nef constructs: SIVmac239 nef, NL4-3 nef and the selected nef chimeras. 48 h post-transfection, cells were analyzed by flow cytometry for autophagosome-associated EGFP-LC3B. Data correspond to the mean and SEM of the percentage of EGFP+ cells from three independent experiments. C, F HEK293T cells were transfected with NL4-3 nef, SIVmac239 nef and the selected chimeras. 48 h later, cells were exposed for 4 h to increasing concentrations of rapamycin (0–4 μM). Next, cells were analyzed by western blot for the levels of GFP, LC3, and ACTB. Densitometric analyses were performed to determine the ratio of LC3-II over LC3-I relative to SIVmac239 nef with no rapamycin treatment. All images are representative of three independent experiments. Significantly different values are indicated by asterisks *P ≤ 0.05; **P ≤ 0.01
In order to narrow down which particular region is responsible for this activity, we generated additional chimeric proteins replacing three portions within the N-terminal domain of NL4-3 Nef, as shown in Fig. 4D. Similar to our previous approach, we first tested the ability of chimeras I.I, I.II and I.III to impair autophagosome formation by measuring saponin resistant EGFP-LC3 using flow cytometry. Whereas chimeras I.I and I.II retained the full potential to limit autophagosome biogenesis, chimera I.III did not exhibit such effect on autophagy (Fig. 4E). Consistent with this, we also found that unlike the chimeras I.I and I.II, the chimeric protein I.III was not able to prevent LC3 lipidation upon autophagy activation by rapamycin (Fig. 4F). These observations indicate that the ability to inhibit the initiation of autophagy maps to a region between amino acids 40 and 57 in the N-terminal domain of NL4-3 Nef.
Residues T48 and A49 in NL4-3 Nef are responsible for counteracting autophagy initiationTo identify the specific residues in Nef required to intersect with the early stages of autophagy, we generated 9 pair-wise alanine-scanning mutants by site-directed mutagenesis comprising positions 40 to 57 (Fig. 5A). Of note, positions that naturally harbor alanine residues were replaced by valine. Next, the mutants were tested for their effect on autophagy initiation. For convenience, this panel of mutants along with wild type NL4-3 Nef was cloned into the expression vector pCI and were tagged with HA to facilitate western blot analyses and microscopy studies. For these assays, HEK293T cells were transfected with each of the 9 mutants, using wild-type NL4-3 Nef and an empty vector as positive and negative controls, respectively. As for Fig. 4, their impact on autophagosome biogenesis was assessed first by flow cytometry. All mutants except construct 48–49 Nef (which harbors T48A and A49V substitutions) were successful at preventing autophagosome formation, which is reflected by a significant reduction in the percentage of autophagosome+ cells (Fig. 5B). Consistent with these findings, mutation of residues 48–49 abrogated Nef’s ability to limit autophagy flux, since when cells expressing this mutant were treated with rapamycin a rapid LC3-I-to-LC3-II conversion was observed (Fig. 5C). We previously demonstrated that HIV-1 NL4-3 Nef prevents early stages of autophagy by enhancing an association between BECN1 (a key protein in autophagy initiation) and its natural inhibitor BCL2 [28]. Hence, we sought to investigate the effect of mutations at positions 48–49 in Nef in the BECN1-BCL2 binding. For this, HEK293T cells were transfected with an empty vector (pCI), NL4-3 Nef or the 48–49 Nef mutant. 48 h later, cells were harvested and BCL2 was immunoprecipitated using a BCL2-specific antibody. The pulldown fraction was next analyzed for the presence of BCL2 and BECN1, as previously described [28]. In agreement with our previous findings, wild type NL4-3 Nef, but not the 48–49 Nef mutant, enhanced the association between BECN1 and BCL2 (Fig. 5D). Hence, these observations demonstrate that the ability of Nef to intersect with autophagy initiation through the BCL2-mediated sequestration of BECN1 maps to residues 48–49 in Nef.
Fig. 5
Nef uses residues 48–49 to prevent autophagosome biogenesis. A Alignment comprising residues 40–57 in NL4-3 Nef. Substitutions introduced in each mutant are indicated in red. B HEK293T cells were co-transfected with EGFP-LC3B and either an empty vector, NL4-3 nef or the selected nef mutants. 48 h post-transfection, cells were analyzed by flow cytometry for autophagosome-associated EGFP-LC3B. C HEK293T cells were transfected with NL4-3 nef or the selected mutants. 48 h later, cells were treated with rapamycin (4 μM) for 4 h and analyzed by western blot for HA, LC3, and ACTB. Densitometric analyses determine the ratio of LC3-II:I relative to NL4-3 nef are shown underneath the blots. D HEK293T cells were co-transfected with BECN1 and either an empty vector, NL4-3 nef or 48–49 NL4-3 nef. 48 h later, cells were harvested and BCL2 was immunoprecipitated. The pulldown fraction was examined for BCL2 and BECN1. Lysates were also analyzed by western blot for BECN1, HA, BCL2, LC3, and ACTB. Densitometric analyses indicate the relative BCL2-BECN1 interaction. E HEK293T cells stably expressing EGFP-LC3 were transfected with an empty vector, NL4-3 nef, a Nef mutant harboring alanine substitutions at the residues responsible for blocking autophagy maturation (36–39 Nef) or 48–49 NL4-3 nef. 48 h later, cells were treated with DMSO or rapamycin (4 μM) for 4 h prior to microscopy visualization of EGFP-LC3-coated autophagosomes. Graph: number of autophagosomes per cell from 15 randomly selected cells. Scale bar: 10 μm. Images are representative of three independent experiments. Significantly different values are indicated by asterisks *P ≤ 0.05; ***P ≤ 0.001; ****P ≤ 0.0001
Previous studies have reported that the ability of Nef to impair autophagosome-lysosome fusion maps to residues 36–39, which resemble the domain that Rubicon uses to block autophagy maturation [28, 31]. To confirm that Nef’s abilities to intersect with autophagy initiation and maturation are genetically separable, we assessed autophagosome biogenesis by fluorescence microscopy. For this, HEK293T cells stably expressing EGFP-LC3 were transfected with HA-GST (as an irrelevant protein), NL4-3 Nef, a Nef mutant harboring alanine substitutions at positions 36–39, or the 48–49 Nef mutant. 48 h later, cells were exposed to rapamycin (4 μM) for 4 h, and autophagosome formation was monitored by LC3 puncta. Consistent with our previous findings [28], wild type Nef significantly reduced autophagosome biogenesis. A similar phenotype is observed with the 36–39 Nef mutant, which is unable to block autophagy maturation. This is expected, since this mutant is still able to intersect with autophagy initiation [28]. By contrast, cells expressing the 48–49 Nef mutant displayed high levels of autophagosomes, reflecting its inability to prevent their generation. However, the level of autophagosomes was higher than that of the HA-GST control, supporting the notion that this mutant retains the ability to block autophagosome–lysosome fusion, causing in turn an accumulation of autophagosomes (Fig. 5E). Quantification of autophagosomes from 15 randomly selected cells for each experimental condition further confirms these observations (Fig. 5E; graph). Hence, these findings demonstrate that the ability of Nef to intersect with autophagy initiation and maturation is genetically separable.
The Nef-mediated block in autophagy initiation prevents Gag redistribution to autophagosomes, consequently increasing virion productionTo determine the relevance of Nef’s effects on counteracting the early stages of autophagy in Gag levels, and thus, virus replication, we assessed the subcellular distribution of Gag in the presence of Nef and the 48–49 Nef mutant. For this, HEK293T cells stably expressing EGFP-LC3 were co-transfected with NL4-3 Δnef and either HA-tagged GST, NL4-3 Nef or 48–49 Nef. 48 h later, cells were treated with rapamycin (4 μM) for 4 h to trigger autophagy, and the co-localization of Gag and LC3 was analyzed by fluorescence microscopy. The degree of Gag-LC3 co-localization was determined by calculating the Pearson’s correlation coefficient. As expected, Gag was mainly distributed at the plasma membrane in the presence of wild type Nef, while it largely localized in LC3-coated autophagosomes in cells expressing HA-GST or the 48–49 Nef mutant (Fig. 6A, B). These findings are consistent with our replication assays in Fig. 1, where Nef expression is associated with higher Gag levels, and further support the idea that by impairing autophagosome formation, Nef prevents Gag from being targeted for autophagy elimination. This hypothesis was verified through particle rescue assays. Here, HEK293T cells were co-transfected with NL4-3 Δnef and either pCI, NL4-3 Nef or the 48–49 Nef constructs. 24 h post-transfection, cells were washed, and the culture media was supplemented with rapamycin (4 μM) or DMSO for 12 h. The percentage of maximal virus production was calculated relative to the levels of virions detected in the presence of DMSO for each transfection condition. Whereas the presence of Nef rescued virion production in the presence of rapamycin, the 48–49 Nef mutant failed at doing so (Fig. 6C). In fact, when analyzing the cell lysates, Gag levels were only fully restored by wild type Nef (Fig. 6D). However, despite its inability to rescue virion release to the levels of wild type Nef under rapamycin treatment, cells transfected with the 48–49 Nef mutant afforded higher Gag expression (Fig. 6D) and virion production than those transfected with the pCI vector (Fig. 6C; 50% versus 33% of maximal virus release, respectively), suggesting that the ability of Nef to restore Gag levels, and consequently virion release, requires Nef-mediated block on both autophagy initiation and maturation.
Fig. 6
Nef uses residues 48–49 to prevent Gag redistribution to autophagosomes, restoring Gag and virion levels. A HEK293T cells stably expressing EGFP-LC3 were co-transfected with NL4-3 Δnef and either HA-GST (irrelevant protein), NL4-3 nef, or 48–49 NL4-3 nef. 48 h later, cells were stimulated with rapamycin (4 μM) for 4 h and autophagosome formation was examined by fluorescence microscopy. B The Pearson’s correlation coefficient for the Gag-LC3 co-localization was analyzed from three biological replicates. Scale bar: 10 μm. Images are representative of three independent experiments. C HEK293T w
留言 (0)