
記住我

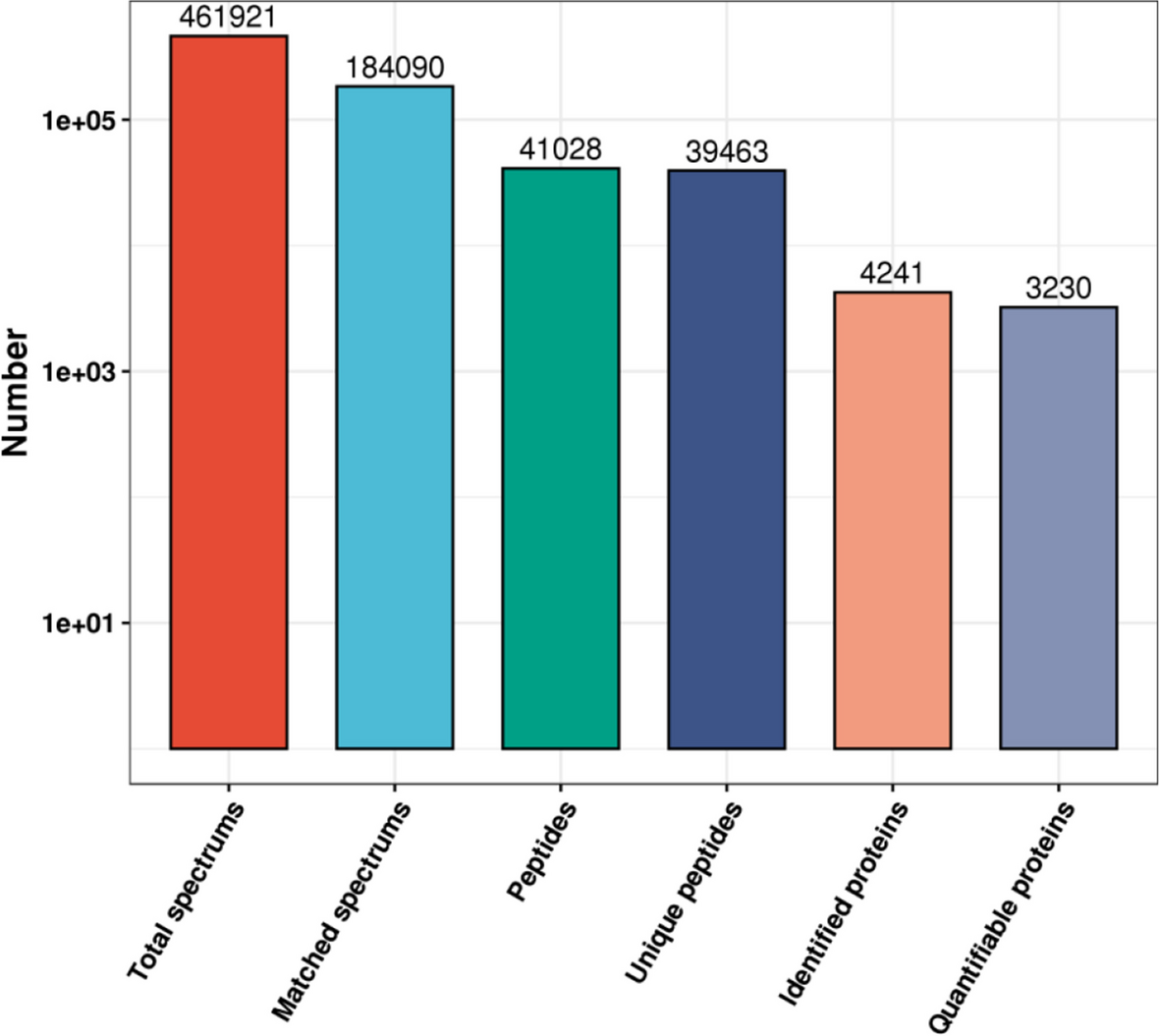






We denote the results with 1% FDR from the reverse, shuffle, pseudo-reverse, pseudo-shuffle, and de Bruijn methods as FDRR, FDRS, FDRPR, FDRPS, and FDRD, respectively.
Saccharomyces cerevisiae datasetWe compared the results for the identified PSMs with the 1% FDR using the S. cerevisiae Elite and 2DLC dataset, the protein database, and various decoy databases. As shown in Fig. 1a and in Supplementary Figure 1b, the numbers of PSMs for FDRR, FDRS, FDRPR, FDRPS, and FDRD were nearly identical regardless of the decoy construction method. (For consideration of the variation in the shuffle, pseudo-shuffle, and de Bruijn method, the results additional databases, in this case four shuffle, four pseudo-shuffle, and four de Bruijn databases, were compared, as shown in Supplementary Figures 1a and b. There was no variation in the shuffle, pseudo-shuffle, and de Bruijn methods.)
Fig. 1
Comparison of the number of PSMs among various databases of typical size and number of target and decoy unique peptides. a, b, c The blue bars show the numbers of PSMs for 1% FDR without the correction factor. The orange bars show the numbers of PSMs for 1% FDR using Factor 2. The gray bars show the numbers of PSMs for 1% FDR using Factor 1. The yellow line show the ratio of target and decoy unique peptide ratio. a The UniProt S. cerevisiae protein database and the S. cerevisiae Elite dataset. b The UniProt human protein database and the HEK293 3-replicate dataset. c The UniProt human protein database and the HEK293 24-fraction dataset. d The blue bars show the ratios of all peptides to unique peptides. The red bars show the ratios of all peptides to redundant peptides. Comparison of the ratio of unique and redundant peptides for the S. cerevisiae and human protein databases
HEK293 datasetsWe used two HEK293 datasets, called the HEK293 3-replicate dataset and the HEK293 24-fraction dataset, and compared the results for the identified PSMs with the 1% FDR among various decoy databases. Figures 1b and 1c show the comparison outcomes for FDRR, FDRS, FDRPR, FDRPS, and FDRD. The numbers of PSMs for FDRR, FDRPR, and FDRD were nearly identical, but the numbers of PSMs for FDRS and FDRPS were, in the HEK293 3-replicate dataset, about 6% lower, and in the HEK293 24-fraction dataset, they were about 8% lower than those for FDRR, FDRPR, and FDRD. Hence, the FDR is overestimated for FDRS and FDRPS, unlike in the S. cerevisiae datasets. Additionally, we used ten cell line datasets and compared the results for the identified PSM with the 1% FDR outcomes among the various decoy databases. Supplementary Figure 2 shows the comparison results for FDRR, FDRS, FDRPR, FDRPS, and FDRD. The numbers of PSMs for FDRR, FDRPR, and FDRD were nearly identical, but the numbers of PSMs for FDRS and FDRPS were about 9% (on average) lower than those for FDRR, FDRPR, and FDRD.
The ratio of unique and redundant peptides in the S. cerevisiae and the human protein databaseWe compared the ratio of unique (non-redundant) peptides in the target database and various decoy databases to analyze the cause of FDR overestimation for FDRS and FDRPS in the HEK293 datasets. Unique peptides were generated with the following parameters: missed cleavage = 2, min length = 2, max length = 45, and NTT = 2. As shown in Figs. 1a and 1b, in the S. cerevisiae protein database, the ratios of unique targets and unique decoy peptides in the reverse database are nearly identical at 49.97:50.03. Shuffle databases have a ratio of 47.19:52.81 on average, the ratios for the pseudo-reverse and de Bruijn databases are 50.05:49.95 and 49.97:50.03 (on average), and the ratio for the pseudo-shuffle database is 48.45:51.55 on average. In the human protein database, the ratio of unique targets and unique decoy peptides for the reverse database is 49.91:50.09, the ratios for the pseudo-reverse and de Bruijn databases are 50.06:49.94 and 49.70:50.30 (on average), whereas shuffle databases have a ratio of 18.02:82.98 on average, and pseudo-shuffle databases show a ratio of 19.19:80.81 on average. (The ratios of another four shuffle databases, four pseudo-shuffle databases, and four de Bruijn database are shown in Supplementary Figure 1).
We found that when the (pseudo) shuffle decoy database is used, the ratios of unique peptides of a target database and a decoy database differ significantly in the human protein database. To find the reason for this, the ratios of redundant peptides in the S. cerevisiae and human target databases were compared. A redundant peptide refers to an overlapping peptide from among all peptides in the target database. For example, when protein A is “ATCDEFRGHIPKLNP” and protein B is “YKLMNWRGHIPK,” the tryptic peptide “GHIPK,” which is common to proteins A and B, is termed a redundant peptide. The redundant peptides of the S. cerevisiae target database amounted to 7.09% of all peptides, and the redundant peptides in the human target database amounted to 77.38% of all peptides (Fig. 1d).
The ratio of redundant peptides has a considerable influence on the ratio of unique peptides when a decoy database is created using the (pseudo) shuffle method. For example, when there are three overlapping peptides “ACDEFG” in the target database, the (pseudo) reverse method creates three identical peptides “GFEDCA”. Because the overlapping peptides are removed, the unique peptide created when the (pseudo) reverse method is used has only one unique peptide in each of the target and decoy databases. However, given that the (pseudo) shuffle method creates three different peptides, such as “FEGDCA,” “AFEDCG,” and “DCAGEF,” the unique peptides created by the (pseudo) shuffle method consist of one unique peptide in the target database and three unique peptides in the decoy database. Eventually, as the ratio of redundant peptides increases, if the decoy database is created using the (pseudo) shuffle method, an imbalance of unique peptides occurs, as shown in Figs. 1a and 1b. As a result, as shown in Figs. 1b and 1c, FDRS and FDRPS have fewer PSMs compared to FDRR, FDRPR, and FDRD.
The correction factor is needed when estimating the FDRWe compared the 1% FDR results with the correction factor proposed by Elias and Gygi (Factor 1) [7] and that by Kim et al. (Factor 2) [14] and without the correction factor. As shown in Fig. 1a and in Supplementary Figure 1b, in the S. cerevisiae protein database, FDRR and FDRPR with the correction factor showed about -0.14 and 0.08% (on average) for the S. cerevisiae Elite dataset, and about -0.07 and -0.12% (on average) for the S. cerevisiae 2DLC dataset more(more less) PSMs compared to those without the correction factor. In addition, FDRS, FDRPS and FDRD with the correction factor showed corresponding increases in the number of PSMs of about 0.89, 0.64 and -0.19% (on average) for the S. cerevisiae Elite dataset and about 0.67, 0.43, and -0.07% (on average) for the S. cerevisiae 2DLC dataset.
As presented in Figs. 1b and c, for the human protein database, FDRR, FDRPR, and FDRD with the correction factor showed increases in the number of PSMs of about -0.06, -0.02, and -0.17% (on average) for the HEK293 3-replicate dataset, and by about 0.0%, 0.0% (identical), and -0.23% (on average) for the HEK293 24-fraction dataset. On the other hand, FDRS and FDRPS with the correction factor showed increases in the number of PSMs by about 7.82 and 7.74% (on average) for the HEK293 3-replicate dataset and by about 12.20 and 12.58% (on average) for the HEK293 24-fraction dataset on average. (The results of another four shuffle databases, four pseudo-shuffle databases, and four de Bruijn databases are presented in Supplementary Figure 1) Additionally, as shown in Supplementary Figure 2, FDRR, FDRPR, FDRD with the correction factor showed increases in the number of PSMs by about -0.12,- 0.08 and -0.14% (on average) for the ten cell line datasets. On the other hand, FDRS and FDRPS with the correction factor showed increases in the number of PSMs by about 12.35 and 12.46% (on average) for ten cell line datasets on average.
These results indicate that FDRR, FDRPR, and FDRD in both the S. cerevisiae and human protein databases showed slight differences regardless of whether or not the FDR was estimated with the correction factor. In the S. cerevisiae protein database, there was little difference between FDRS (and FDRPS) with the correction factor and FDRS (and FDRPS) without the correction factor. However, in the human database, when FDRS (and FDRPS) with the correction factor and FDRS (and FDRPS) without the correction factor were compared, we found that the number of PSMs for FDRS (and FDRPS) with the correction factor increased significantly. In other words, in the human protein database, if the FDR was estimated using the (pseudo) shuffle database without the correction factor, it was overestimated. Accordingly, it is important to estimate the FDR with the correction factor.
The S. cerevisiae and the human six frame translation databaseWe used 6FT databases to analyze the degree of FDR overestimation for FDRS and FDRPS in large databases. First, the ratio of unique peptides in the target database and various decoy databases is compared for 6FT databases. Unique peptides were generated with the following parameters: missed cleavage = 2, min length = 2, max length = 45, and NTT = 2. As shown in Fig. 2, in the S. cerevisiae 6FT database, the ratio of unique targets and unique decoy peptides in the reverse, pseudo reverse and the de Bruijn databases are nearly identical at 50.03:49.97, 50.04:49.96, and 49.72:50.28, respectively. For S. cerevisiae, the shuffle and pseudo shuffle databases have ratios of 25.67:74.33 and 27.30:72.70, respectively. In the human 6FT database, the ratio of unique target and unique decoy peptides in the reverse, pseudo reverse and de bruijn database are nearly identical at 50.45:49.55, 50.45:49.55, and 50.24:49.76, respectively. The shuffle and pseudo shuffle databases have corresponding ratios of 44.02:55.98 and 45.67:54.33.
Fig. 2
Comparison of the numbers of PSMs of various databases of large size (six-frame translation). The blue bars show the numbers of PSMs for 1% FDR. The orange bars show the numbers of PSMs for 1% FDR using Factor2. The gray bars show the numbers of PSMs for 1% FDR using Factor 1. The yellow line show the ratio of target and decoy unique peptide ratio. a The S. cerevisiae six frame translation protein database and S. cerevisiae Elite dataset. b The human six frame translation protein database and the HEK293 3-Replicate dataset. a The S. cerevisiae six frame translation protein database and S. cerevisiae 2DLC dataset. b The human six frame translation protein database and the HEK293 24-Fraction dataset
As shown in Supplementary Figure 3, these results indicate that FDRR, FDRPR, FDRD in both the S. cerevisiae and human protein databases showed slight differences regardless of whether or not the FDR was estimated with the correction factor. However, in the S. cerevisiae 6FT database, FDRS and FDRPS with the correction factor showed corresponding increases in the number of PSMs by about 9.81 and 9.90% (on average) for the S. cerevisiae Elite dataset, and by about 7.88 and 8.29% (on average) for the S. cerevisiae 2DLC dataset. In the human 6FT database, FDRS and FDRPS with the correction factor showed corresponding increases in the number of PSMs by about 1.62 and 1.10% (on average) for the HEK293 3-replicate dataset, and by about 2.22 and 1.73% (on average) for the HEK293 24-fraction dataset.
In addition, we ran a comparison using the separate FDR [15] which is often used in proteogenomics. For the separate FDR, we divided known databases and novel databases. It is easy to divide known and novel databases for the reverse, pseudo reverse, De bruijn decoy databases, but not for the shuffle and pseudo shuffle decoy databases. We note that the known database is the UniProt database and its decoy database and the novel database is the 6FT database apart from the known database. In addition, we calculated the correction factor using these known and novel databases. As shown in Supplementary Figure 3 and Supplementary Table 2, FDRR, FDRPR and FDRD of the S. cerevisiae and human protein databases show the same number of PSMs, even if the correction factor is used. In the S. cerevisiae 6FT database, FDRS and FDRPS with the correction factor showed corresponding increases in the number of PSMs by about 0.93 and 0.31% in known PSMs and by about 35.00 and 8.70% in novel PSMs for the S. cerevisiae Elite dataset, also showing corresponding increases in the number of PSMs of about 0.77 and 0.46% in the known PSMs and of about 11.24 and 81.13% in the novel PSMs for the S. cerevisiae 2DLC dataset. In the human 6FT database, FDRS and FDRPS with the correction factor showed corresponding increases in the number of PSMs by about 5.82 and 6.00% in the known PSMs and by about 0.03 and 0.00% (identical) for the novel PSMs for the HEK293 3-replicate dataset, and showed increases in the number of PSMs by about 12.32%, 13.07% in known PSMs and by about 2.71%, 0.00% (the same) in novel PSMs for the HEK293 24-fraction dataset. We used a simple method to divide known and novel databases. However, it is likely difficult to divide known and novel databases for the separate FDR. In proteogenomics for the separate FDR, we do not recommend the use of the shuffle and pseudo shuffle decoy databases, because it is difficult to divide known and novel database.
留言 (0)