
記住我









The integrated stress response (ISR) is an evolutionarily conserved mechanism to cellular stress that is activated by different intrinsic and extrinsic factors. The main function of the ISR is to maintain cellular homeostasis by downregulating protein synthesis and upregulating specific target genes [1]. The key intrinsic stress is endoplasmic reticulum stress (ER stress), which occurs when the capacity of protein folding is exceeded [1, 2]. Extrinsic stressors include glucose and amino acid deprivation, hypoxia, viral infections, and the presence of reactive oxygen species (ROS) [1]. The ISR works with several other cellular adaptation pathways such as proteotoxicity, ubiquitin–proteasome, autophagy, phosphatidylinositol‐3 kinase (PI3K) signaling, and unfolded protein response (UPR); which act in a time-dependent manner upon induction of any stress mediated signaling [3,4,5,6,7,8,9,10]. Moreover, the cross-talk of ISR with osmotic stress response (OSR), DNA damage response (DDR), and heat-shock protein (HSP) response is reported and is often known to function in a cytoprotective manner [11,12,13,14].
There are four regulatory ISR-kinases, all of which converge on the phosphorylation of the alpha subunit of the eukaryotic initiator factor 2 (eIF2α) at the serine site 51 [15]. Phosphorylation of eIF2α reduces global protein synthesis and the formation of stress granules are also reported in some cases, while promoting the translation of stress-related genes such as activating transcription factor 4 (ATF4) [16], which supports cell survival. However, under conditions of severe cellular stress, the adaptive response loses its ability to effectively alleviate the stress and instead promotes the activation of apoptotic pathways [17, 18]. The ISR is terminated with the dephosphorylation of eIF2α in a negative feedback loop manner. The ISR has been observed to be activated in different neurodegenerative disorders like multiple sclerosis (MS), Alzheimer’s disease (AD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis (ALS) [19]. This review outlines ISR mechanisms, and its relevance in CNS homeostasis and in neurodegenerative disorders where ER stress and ISR link together to contribute to neuronal cell death, inflammation, protein aggregation, and cognitive impairment. We will also provide an overview on the current therapeutic strategies targeted toward the modulation of dysregulated ISR in neurodegenerative disorders.
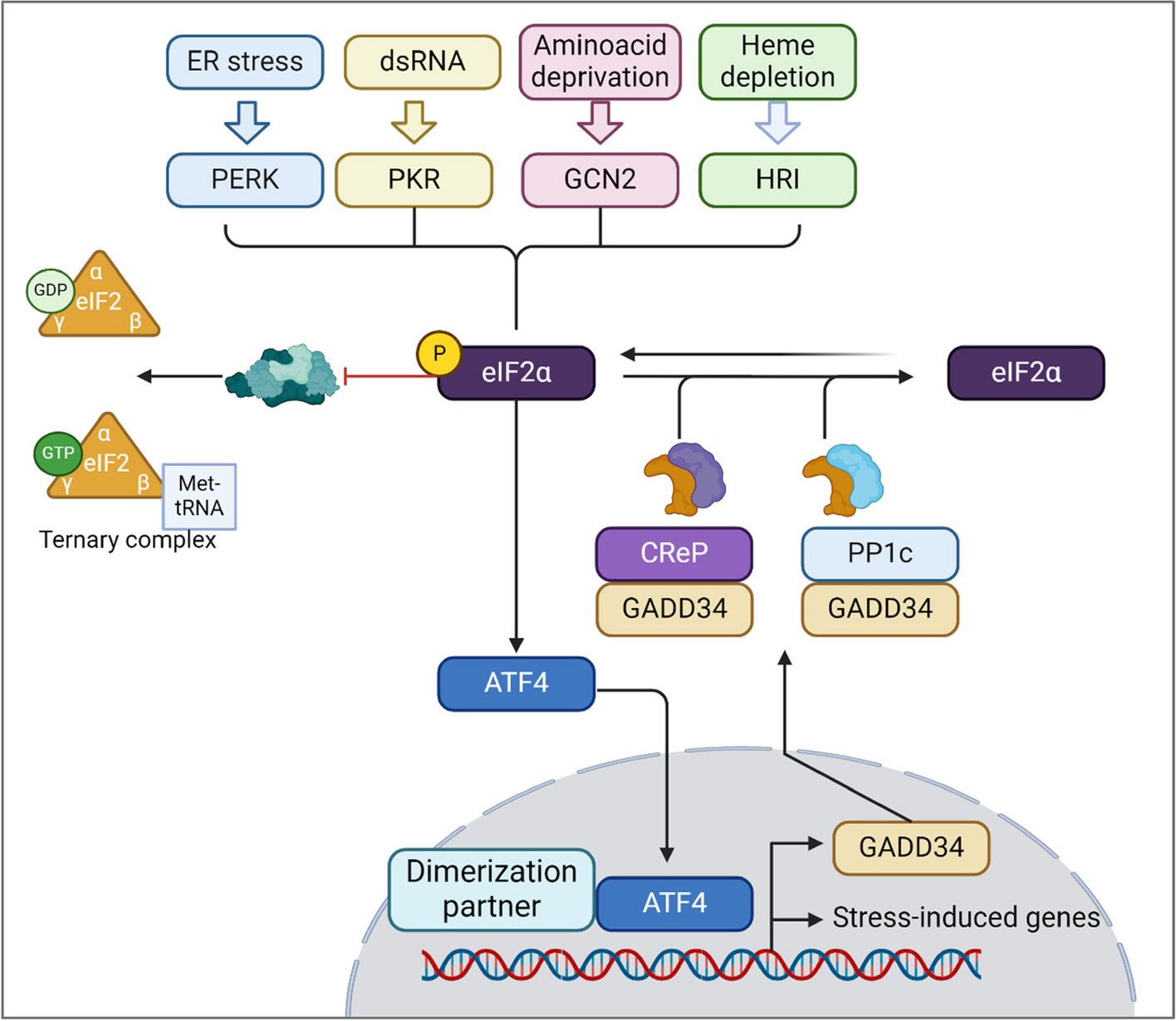
Initiation of the ISR: Four sensors, one phosphorylationThe ISR is first initiated by a disturbance in homeostasis and is recognized by the eIF2α protein kinases (EIF2AKs). When changes in homeostasis are detected, the kinases phosphorylate the alpha subunit of the eukaryotic initiator factor 2 (eIF2α) to inhibit protein synthesis, reconfigure gene expression for stress adaptation or inducing apoptosis [1]. The four kinases are PKR-like ER kinase (PERK), protein kinase double-stranded RNA dependent (PKR), general control non-derepressible-2 (GCN2), and heme-regulated inhibitor (HRI) [15, 20]. While each kinase has its own regulatory mechanisms, they all converge on the phosphorylation of eIF2α at the serine site 51 (Fig. 1), which is critical for translation control [21, 22]. PERK, also known as EIF2AK3, is an ER transmembrane protein that becomes activated upon detecting disturbances in the ER, like the accumulation of misfolded or unfolded proteins, calcium depletion, or redox imbalance [1, 23, 24]. There are two proposed models for PERK activation. One model suggests that, in homeostatic conditions, PERK remains inactive as its luminal domain is bound to GRP78. Upon ER stress, GRP78 dissociates from PERK to bind with unfolded proteins, leading to PERK dimerization and autophosphorylation that initiates downstream signaling [9, 25, 26]. The second and most recent model suggests an alternative mechanism in which PERK can be activated directly by binding to unfolded or misfolded proteins via its luminal domain [27, 28]. Once activated, PERK then phosphorylates eIF2α (p-eIF2α), preventing the translation of mRNA and reducing protein synthesis, which allows the ER to either refold or dispose of the misfolded proteins [15]. Simultaneously, p-eIF2α initiates the translation of ISR-specific mRNAs, like ATF4 [1].
Fig. 1
The integrated stress response signaling pathway. The ISR can be initiated upon sensing ER stress, dsRNA, amino acid deprivation, and heme depletion, which are recognized by PERK, PKR, GCN2, and HRI respectively. Once activated, the four kinases converge in the phosphorylation of eIF2α that then activates ATF4. ATF4 translocate to the nucleus in which promotes the translation of stress response genes. Evidence shows regulation of ISR through “negative feedback”. ATF4 induces the expression of GADD34, which promotes the dephosphorylation of eIF2α by recruiting CReP and PP1c, hence restoring protein synthesis. Figure was created in BioRender
Unfolded proteins are not the only trigger for PERK autophosphorylation. It has been reported that saturation of lipids and modifications in the lipid composition of the ER membrane can trigger PERK activation [29]. Moreover, early studies reported that glucose deprivation in cultured hippocampal neurons leads to PERK activation and an increased expression of caspase-12, an ER resident caspase that has been associated with stress-induced apoptosis [30]. PKR (EIF2AK2) is found in the cytosol and nucleus of mammalian cells [1, 15, 31]. In addition to mounting an interferon response to viral infection [3], PKR also responds to other stimuli such as metabolic and ER stress [31, 32], as well as oxidative stress [33]. Upon stress, PKR dimerizes and auto-phosphorylates, forming a PKR-PKR complex that phosphorylates eIF2α [15]. Studies have shown that prolonged activation of PKR after oxidative stress increased the sensitivity to apoptosis, making its downregulation important for promoting cell survival [33, 34]. Importantly, PKR induces activation of the pro-apoptotic factor C/EBP homologous protein, CHOP, in response to hyperoxia [35]. GCN2 (EIF2AK4) is proposed to regulate changes in gene expression due to acid and glucose deprivation by sensing uncharged transfer ribonucleic acid (tRNA), although the exact mechanism is yet to be elucidated [15, 36]. Under amino acid deprivation, tRNA accumulates in the A site of the ribosome where it is recognized by the GCN2 regulatory domain histidyl-tRNA synthetase (HisRS), triggering GCN2 dimerization, eIF2α phosphorylation, and ISR activation [4]. GCN2 can also sense other stresses. For instance, inhibiting the proteasome system leads to the formation of stress granules that are primarily recognized by GCN2 and initiates the ISR [37, 38]. HRI or EIF2AK1 is mainly found in erythroid cells. Early studies reported that HRI is involved in erythrocyte differentiation and is required to produce α and β globin in red blood cell (RBC) precursors and it also promotes the survival of these cells under heme deficiency [39]. When the availability of iron decreases, eIF2α phosphorylation by HRI inhibits the translation of globin mRNAs, prevents hemoglobin production and exerts protection against toxic globin aggregates [1, 39]. HRI has also been found to initiate autophagy in cases when α-synuclein is overexpressed, hence helping to clear out the protein aggregates accumulated in the cytosol [40]. It is also demonstrated that HRI is required for inflammatory responses during infection [41]. Recent studies show that HRI acts as a proteotoxicity sensor via a pathway involving Hsp70, Bag3 and HRI, which detects the abnormal accumulation of proteins in the cytosol and triggers the phosphorylation of eIF2α [42]. All four kinases phosphorylate eIF2α at its serine site 51 and with this in common, they can often overlap and act cooperatively to sense different stress stimuli and integrate them to achieve specific cell responses, hence the name “integrated stress response”.
Downstream p-eIF2a, cellular responses, and termination of the ISRUnder homeostatic conditions, eIF2 participates in mRNA translation and recognition of the initiation codon AUG [43]. eIF2 is a 126 kDa heterotrimer protein comprised of α, β and γ subunits [44] with the eIF2α subunit playing a major regulatory role due to its RNA binding and phosphorylation sites [1]. Translation initiation involves the assembling of elongation-competent 80S ribosomes with an initiator tRNA at the ribosomal P site 1. This two-step process requires at least 9 eukaryotic initiation factors including eIF2. The first step requires the formation of 48S initiation complexes, which then join 60S subunits [43]. Many of the 48S initiation complexes are formed by a 43S preinitiation complex which is comprised of a 40S subunit, the eIF2-GTP-Met-tRNAiMet ternary complex, and other eIFs like eIF3, eIF1, eIF1A, and eIF5. This complex is composed of eIF2, guanosine triphosphate (GTP), and charged methionyl-transfer RNA (Met-tRNAiMet) [43, 45]. Upon stress, p-eIF2α alters the regular translation initiation by inhibiting the formation of active GTP from the eIF2-GDP complex and binds strongly to a modulatory portion of eIF2β that inhibits the formation of active GTP from the eIF2-GDP bound form. Consequently, there is less availability of the ternary complex, which leads to decreased translation rates (Fig. 1) and increased translation of ISR-related mRNAs like ATF4, ATF5, CHOP, and GADD34 [1, 45].
ATF4 is a key regulator in the ISR network, as it is vital for relieving ER stress by either promoting adaptation or triggering apoptosis [1]. ATF4 downstream activity initiates with the formation of homo and or heterodimers with other basic leucine zipper (bZIP) transcription factors including CHOP or AP-1 members that are known to regulate the transcriptional selectivity and thereby influence the outcome of ISR [46, 47]. The interacting heterodimers bind to cAMP (cyclic adenosine monophosphate) responsive elements to control target gene expression [18], which leads to the transcriptional upregulation of stress-related genes and pathways related to amino acid transport and metabolism as ATF4 can promote cell survival by inducing autophagy. Following amino acid starvation and ER stress, GCN2 and PERK along with ATF4 and CHOP can promote the expression of genes related to the autophagosome formation and function [48]. Dimer combinations between ATF4 and CHOP can regulate transcription through various mechanisms. In response to leucine starvation, ATF4-CHOP heterodimers regulate genes related to the degradation of ubiquitinated substrates, such as Nbr1, Atg7, and p62 [48]. Autophagy-related genes Atg10, Gabarap, and Atg5 are expressed after the formation of ATF4-CHOP heterodimer in response to amino acid deprivation [48]. Other reports suggest that ATF4 alone can target genes related to amino acid transport and biosynthesis, and it can act along with CHOP to regulate several shared genes related to protein synthesis, mRNA translation, and the unfolded protein response (UPR) [17]. The UPR is a signaling pathway that becomes activated due to ER stress and detects unfolded proteins through ER transmembrane receptors: PERK, IRE1, and ATF6. Both the ISR and UPR converge in the phosphorylation of eIF2α through PERK (Fig. 2) [1]. eIF2α phosphorylation can either promote cell survival or cell death. The increase in protein synthesis mediated by ATF4 and CHOP can promote cell death by oxidative stress and depletion of ATP [1, 18, 49]. This dual role of ATF4 has been attributed to the formation of heterodimers with different binding partners that lead to different responses. Binding partners of ATF4, such as C/EBPβ and C/EBPγ are involved in signal adaptation, upregulation of stress response genes, and protection against oxidation [50, 51]; while ATF4-CHOP is mainly associated with autophagy and pro-apoptotic activity. As reviewed previously by others [52], CHOP has been identified to induce apoptosis; however, as described earlier, it can also promote cell survival. This discrepancy may reflect the duration and level of stress, as well as the level of CHOP expression. These findings suggest that ATF4/CHOP may promote an initial survival response; however, with prolonged stress CHOP expression will initiate cell death to restore homeostasis [52].
Fig. 2
Mitochondrial dysfunction and ISR: The activation of the integrated stress response (ISR) during mitochondrial dysfunction is triggered by various mechanisms. The fragmentation of mitochondrial DNA is among the leading causes of mitochondrial dysfunction, which is managed by an ISR sensor, protein kinase RNA-activated (PKR). Mitochondrial dysfunction during amino acid metabolism is managed by another class of ISR kinase- general control nonderepressible-2 (GCN2). The Tri-Carboxylic Acid (TCA) cycle is fed via the amino acid degradation during metabolic rewiring stage of mitochondrial stress, and the depletion of amino acids results in activation of GCN2. During this process, the equilibrium maintenance of reducing equivalents is maintained by the malate and aspartate shuttle. Upon induction of mitochondrial stress by generation of reactive oxygen species (ROS), a mitochondrial protease known as OMA1 regulates the mitochondrial stress dynamics by cleaving DAP3-binding cell death enhancer 1 (DELE1). This subsequently activates heme-regulated inhibitor (HRI) after translocating to the cytoplasm. The crosstalk between mitochondria and endoplasmic reticulum aids ER to sense the alterations in the levels of calcium (Ca2 +), ROS, and changes in energy productions; leading to activation of ER transmembrane protein- PKR-like ER kinase (PERK). Upon mitochondrial dysfunction and the ER-mitochondrial crosstalk, these protein kinases phosphorylate eIF2α and inhibit global protein translation to mitigate stress and restore the normal homeostasis. Until the stress is resolved, a translational shift due to upstream open reading frame (uORF) mediates selective translation of proteins such as ATF-4 and usurps the global translation to mitigate the stress. ATF-4 along with its other dimerization partners regulates amino acid synthesis genes, antioxidant pathways, chaperones and metabolism related genes to restore the homeostasis post stress. However, chronic stress results in activation of CHOP-mediated apoptotic cell death. Figure was created in BioRender
The termination of the ISR, restoration of protein synthesis, and return to homeostasis occur upon eIF2α dephosphorylation in a process regulated by the protein phosphatase 1 (PP1) complex through its catalytic subunit (PP1c). The phosphatase activity can be regulated by GADD34 (growth arrest and DNA damage-inducible protein 34) or by the “constitutive repressor of eIF2α phosphorylation”, known as CReP [53, 54]. CReP forms a complex with PP1c to enable homeostasis by maintaining low activity of p-eIF2α, whereas GADD34 expression is induced during the late stages of the ISR in response to ATF4 and forms a complex with PP1 to promote eIF2α dephosphorylation. Thus, this negative feedback loop appears to be essential for restoring homeostasis after a stress response [1, 53].
ISR and mitochondrial UPRMitochondrial stress responses (MSRs) can also result in the buildup of misfolded or damaged proteins. Mutations or deletions in mitochondrial DNA (mtDNA) causes an accumulation of unfolded proteins, which can trigger the activation of the mitochondrial unfolded protein response (UPRmt) [55]. Growing evidence has implicated ISR in UPRmt [56, 57] (Fig. 2). Studies in S. cerevisiae and C. elegans indicate that UPRmt is regulated by ISR sensors or eIF2α kinases. GCN2 depletion is known to significantly upregulate the expression of mitochondrial chaperones that activates UPRmt [55]. Another study revealed that during mitochondrial stress, activation of HRI, even with the absence of full heme, attenuates the global protein translation through phosphorylation of eIF2α, thus implicating a cross-talk between ISR and mitochondrial dysfunction [58]. During the active state of ISR and upon diverse mitochondrial insults, the upstream open reading frames (uORF’s) are involved in translation of selective transcription factors such as ATF4, ATF5 and CHOP [59,60,61]. The accumulation of misfolded or unfolded proteins in the mammalian mitochondrial matrix often leads to the upregulation of chaperones in mitochondria but no ER stress protein response is initiated [61]. Interestingly, UPRmt activation also leads to a reduction in the transcription of oxidative phosphorylation (OXPHOS) components, as mitochondria attempt to lessen their functional demands while addressing the stress [
留言 (0)