In this study, we investigated the mutational burden in ASD and other neurodevelopmental genes among macaques in two fundamental ways. First, we determined whether genes associated with ASD and a number of related neurodevelopmental disorders in humans show evidence of evolutionary constraint against the accumulation of mutations. Second, we examined whether any of those macaque mutations that are predicted damaging in humans overlapped with known ASD and other NDD-associated mutations in humans. Our analysis identified evidence of evolutionary constraint against the accumulation of mutations across all ASD and other NDD gene sets, indicating that, in macaques, as in humans, these genes have important functions. We also demonstrated that predicted damaging mutations were still quite common in all NDD genes, including those that overlap across all NDD disorders and are presumably the most important. Again, given their predicted damaging nature, it might be anticipated that these variants would have phenotypic consequences. Warren et al. [24] examined sequence diversity in a sample of 853 macaques and identified thousands of missense variants across the genome impacting genes associated with human disease. Our results are largely consistent with their study, which analysed sequence diversity in this same dataset. Unlike our study, which set out to examine constraint across neurodevelopmental genes in different ways, theirs specifically examined the frequency of missense variants in these genes compared to all genes and found them to be depleted for missense variants. Nevertheless, our investigation did identify the fact that these NDD genes do harbour missense and other potentially damaging variants that could have phenotypic consequences, as discussed subsequently.
We also demonstrated that, despite constraint, macaques harbour predicted damaging mutations in key NDD genes. These predictions are based on mGAP’s own annotation derived from multiple sources of information after variants are lifted to the human genome, including established prediction tools (such as CADD and SIFT) and ClinVar annotations. These were supplemented with SnpEff annotation for predicted impact on protein-coding genes. Given this, in humans these variants would be expected to have phenotype consequences, although both variable expressivity and variable penetrance will confound the prediction of phenotypic consequences. Recent macaque studies have also shown that macaques with single gene editing or naturally-occurring mutations present a combination of phenotypic behaviours including social and learning impairment as well as repetitive behaviours [19, 21, 32, 33].
One further insight relates to the observation that a number of genes with strong evidence of constraint in humans were not similarly constrained in macaques. Some of these genes, such as CACNA1D, MBD5, AUTS2 and NRXN1 are key neurodevelopmental genes with high penetrance for clinical phenotypes. For example, copy number variation in NRXN1, which encodes a synaptic scaffolding protein, has a penetrance of 6.4% for schizophrenia and 26% for ASD or ID [34]. MBD5, a transcriptional regulator is implicated in ID, ASD, epilepsy and specific cognitive impairments [35]. CACNA1D, which encodes one of the L-type calcium channels is similarly associated with (SFARI Category 2) ASD, ID and epilepsy. Crucially, these genes encode important aspects of brain structure and function, so their constraint against mutations in humans, and the phenotype consequence contingent on mutations occurring, is unsurprising. It is curious, therefore, that similar constraint is not seen in macaques. Gao et al. (2023) also identified a small number of variants with strong evidence of pathogenicity in humans that appeared to be well-tolerated in NHPs, proposing that interactions within the genomic neighbourhood may be relevant. Another possibility is that constraint is more fine-grained than at the gene level. For example, previous research has examined patterns of constraint among Pfam protein domains, showing tolerance to be consistent across specific domains in different genes, and that evolutionary conservation is correlated between entropy measured from Pfam and ExAC [36]. Moreover, among some ID-associated genes, pathologic de novo mutations have been shown to cluster in specific protein domains [37]. Examining patterns of constraint in different ways among primate datasets may therefore offer further insight into pathophysiology in human NDDs.
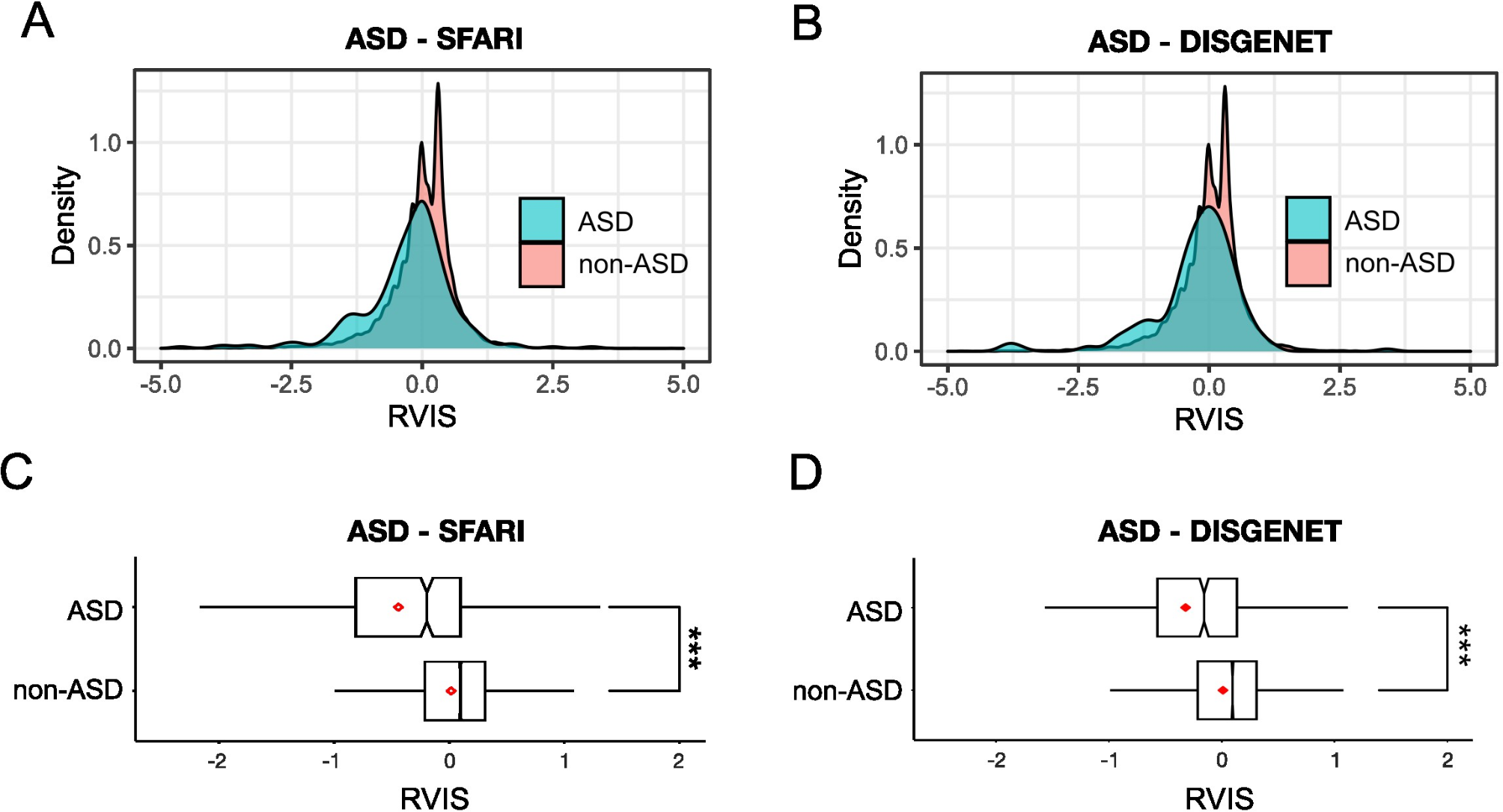
Our decision to use the RVIS was based on our belief that it is a well-established measure of constraint, used across species, that provides an easily interpretable metric for readers. Another widely used metric is the pLI, which uses expectation-maximization to estimate intolerance to loss of function (LoF) variation specifically. Given the exploratory nature of this study, we were interested in casting the net wide beyond LoF to also include missense mutations. We also aimed to use a metric that would be straightforward to interpret and could inform future NHP studies investigating phenotypes in constrained NDD genes, as well as genes that demonstrate constraint in humans but not NHPs. Notably, discrepancies can arise between different methods used for measuring constraint. For example, we identified CNTNAP2 as highly constrained, while gnomAD assigns a pLI of 0 to this gene, suggesting its tolerance to mutation. This anomaly can be attributed to several factors, primarily the specific population studied, the availability and interpretation of phenotypic data, and the relative lack of functional data [38]. It is also expected that metrics will be revised over time as more data accumulate. In clinical situations, it is essential that these metrics are not interpreted in isolation; however, in the research, they provide valuable insights into which genes should be prioritised. This is especially important in NHP research, which can be very costly to undertake.
Although our correlational analysis of human and macaque genes was significant, the value of 0.39 is lower than expected. Supplementary Fig. 1 illustrates the most of genes are clustered around the mean, and it may be the more peripheral genes that are driving this lower correlation. Notably, there are a small number of genes whose scores do not align in humans and macaques and these may be worthy of further investigation (examples include DNAH10, TTN). Additionally, given that the accumulation of mutations and its converse, are dynamically established through evolution, these differences may be indicative of the true impact of evolution on constraint across species, and may potentially provide insights into species-specific phenotypes.
Key differences at a transcriptomic [39] and cellular [40] level have also been observed across human and NHP species consistent with shared and unique dynamic developmental processes. In particular, the retention of duplicated genes is widespread across species, and likely plays a role in determining evolutionary pathways. The macaque reference genome has good coverage for segmental duplications (SDs) [24] that correspond to recent expansion in the major histocompatibility complex (MHC) and other gene families in this species. Although the frequency of SDs approximates humans, in macaques these SDs are more clustered. It is possible that the duplication of genes may result in copies with variable constraint.
Human studies have identified many genes associated with these NDDs; however, much is still not known about the neurophysiological, cognitive and other intermediate phenotypes. There are several limitations to human studies that may be overcome by shifting the focus to NHP-based research. One challenge has been the involvement of less able individuals with ID, ASD or epilepsy, who may be unable to follow instructions or comply with neuroimaging and electrophysiology, which are important tools for studying NDDs. It is important that such individuals are included in research, as they often represent the ‘purest’ forms of the disorder among whom there is wide consensus regarding diagnosis. However, recruiting such individuals and conducting research in an ethical manner has also been difficult, given the restrictions inherent in any research protocol.
New models are therefore needed. Up to now, NDDs have largely been studied in rodent models. However, rodents and humans have diverged significantly more than 70 million years ago, there are major differences in brain structure and function between the two species. This makes it difficult to recapitulate the research paradigms that capture clinical phenotypes for NDDs in rodents. NHP models offer many advantages over rodent models for studying NDDs. Macaques are a particularly promising model for studying NDDs as they share many similarities with humans in terms of social behaviours and have a well-characterised genome. Given that macaques are among our most recent NHP ancestors, we might anticipate that genes that are important in humans would also be important in macaques. It is therefore perhaps unsurprising that NDD genes show evolutionary constraint similar to humans. These genes clearly play a key role in core brain functions, as evidenced in our network analysis, and as such are presumably central to developmental brain processes. Moreover, given that the NDDs studied are all characterised by language abnormalities (including language pragmatics) and that humans have more advanced vocal communications than macaques, there may be reasons to predict that these genes might be associated with more fundamental vocal communication or language systems with a shared a common evolutionary origin. Indeed, recent studies have revealed evolutionarily conserved abilities [41, 42] and brain signatures [43,44,45] associated with language precursor in both humans and macaques. Given our findings, the role of these genes in brain development is more likely to be pre-linguistic and hence at a far more fundamental level than the traits that characterise these disorders. Consequently, these genes may play a less important role in the human specific ‘high-level’ manifestations of these disorders, which include aspects of social skills and language itself. What ultimately determines the clinical phenotype itself is downstream of the genes through their interaction with molecular neurobiology, neural circuits and connectivity through environment and other, as yet undetermined, factors.
Furthermore, the recent availability of a reference genome, a spatial transcriptomic atlas of the macaque cortex [46] enables cross-species comparison of humans and macaques in specific brain regions. However, a hypothesis is still needed to guide the selection of regions of interest associated with phenotypic behaviours. The recent comparative macaque and human studies have identified evolutionarily conserved cognitive systems within the prefrontal cortex [43,44,45], with the cognitive functions hypothesised to be impaired in ASD [47,48,49]. Additionally, human and macaque transcriptomic studies, including recent advances in single-cell genomics and spatial transcriptomic technologies, have revealed enriched gene expression of ASD-associated genes in the prefrontal cortex [50].
Furthermore, the usefulness of macaque Social Responsiveness Scales (mSRS) [15, 16], which is a screening questionnaire that can identify animals who demonstrate vulnerabilities indicative of ASD based on the SRS that is widely used clinically as a screening tool [51], has been illustrated by several studies, for example, significant cerebrospinal fluid (CSF) arginine vasopressin (AVP) was reported in low-social macaques and also in individuals with ASD [52]. Associations between social behaviours and whole genome sequence data in macaques have also been demonstrated [33].
This highlights the importance of developing a macaque model of ASD and the challenge now is to develop research paradigms that capture the characteristics of neurodevelopmental disorders in both clinical terms and in relation to the neurobiology of cognition that are immediately downstream of these clinical manifestations. Indeed, there is a burgeoning research examining social and communicative skills and aspects of behaviour and temperament among NHPs that will have implications for interpretation of data. For example, one study using heritability estimates has identified a preferential paternal transmission of social skills among male offspring [53], and another has established the heritable nature of anxious or inhibited temperament in multigenerational macaque families [54]. These studies demonstrate the ability to employ similar strategies to those that have been employed with human data, and, crucially, using phenotype measures and phenomenological discussions that overlap very closely with humans. ‘Translating’ the human characteristics of disorders such as ASD into animal behaviour is not straightforward, however, and it is hoped that further work in this area will now thrive in anticipation of the major contribution of NHPs in biomedical science.
Limitations
The lack of behavioural and other phenotypes in the macaques studied limits the conclusions that can be drawn regarding the possible consequences of the pathological mutations described. Furthermore, other measures of evolutionary constraint are available, although research supports consistency between these different metrics. There are also limitations with gene lists used for particular disorders, given that these lists change over time with the accumulation of new evidence. It might be argued that these lists are overly inclusive, especially as we did not set an arbitrary threshold for inclusion but instead chose to include all associated genes in these lists. Given the exploratory nature of our study, this was a deliberate decision. To address potential over-inclusivity, we conducted analyses on genes overlapping between disorders (NDD genes), a list of about 100 genes among which are many of the most robust ASD-implicated and NDD genes.




留言 (0)