
記住我

The patients were evaluated in Qilu hospital of Shandong University. Detailed clinical features as well as family history were obtained from affected individuals and reviewed by a group of neurologists. DNA extracted from the patients and healthy members in the family underwent third-generation sequencing and sanger sequencing. Nanopore RNA sequencing was carried out using patient’s tissue obtained from muscle biopsy. All participated family members were enrolled after obtaining the written consents.
RNA extraction and reverse transcription PCRTotal RNA was extracted from patient’s muscle tissue according to standard protocol. PCR assay was carried out as previously described [8]. Forward primer: 5’-AGGCCTGAATTCCAGTTGTG-3’, reverse primer: 5’-TGTTGGCATCCAGATGTAGG-3’.
Western blot analysisA slice of patient’s muscle tissue was lysed by radioimmunoprecipitation assay lysis buffer containing 1% protease inhibitor. Details of immunoblotting assay were previously described [8]. Antibodies for EPM2A was purchased from Abclonal (A7007).
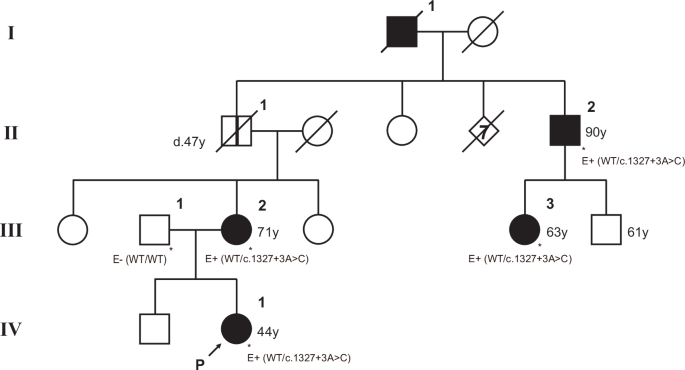
Clinical presentationA 16-year-old girl was admitted to our hospital due to epileptic seizures for three years. At the age of 13, she had a sudden loss of consciousness for several minutes. A few months later, she developed generalized tonic-clonic seizure and was prescribed with antiepileptic drugs which apparently alleviated her symptoms. However, seizures became more frequent at the age of 16 and she gradually developed lethargy, fatigue, headache, cognitive dysfunction, gait disturbance and vision impairment. The above symptoms gradually aggravated despite treatment of antiepileptic drugs. For family history, the patient’s parents were asymptomatic but her younger brother and two cousin sisters had epileptic seizure (Fig. 1A). Her younger brother (III4) suffered from infrequent epileptic seizures since the age of 10. As for III7 and III8, who had the same grandparents with the proband, also presented with epileptic seizures at teenage.
Fig. 1: Family pedigree and verification of the effect of the c.476+14860 C > A variant.
A Pedigree of the family. Black arrow indicates the proband. Diagonal lines indicate deceased individuals. M1 refers to EPM2A c.301+1 G > A. M2 refers to EPM2A c.476+14860 C > A. B The skin biopsy revealed characteristic periodic acid-Schiff (PAS) -positive glycolike intracellular inclusions (Lafora body, LB) in the cytoplasm of a small number of sweat gland ductal epithelial cells. C H&E staining of skin biopsy showed eosinophilic light-stained vacuoles. LBs are pointed by red arrows. D Sanger sequencing using proband’s muscle tissue revealing separated junctions upstream and downstream of EPM2A c.476+14860 C > A. Red arrow indicated the mutation site of EPM2A c.476+14860 C > A. E Reverse transcription PCR showing a shorter PCR product in patient. F Western blot showing absence of EPM2A proteins in patient’s muscle tissue
On examination, the patient responded slowly with an ataxic gait but was unable to complete heel-knee-shin and finger-to-nose testing due to poor cognition. She scored 8 in Mini-mental state examination (MMSE). Video-electroencephalogram (VEEG) showed slow background rhythms and generalized irregular spike and wave discharges. Her visibility decreased to less than 20 cm in both eyes. Fundus photograph and optical coherence tomography were unremarkable. Brain magnetic resonance imaging was normal. Right axilla skin biopsy showed the presence of intracytoplasmic inclusion bodies at the eccrine duct (Fig. 1B) with positive Periodic acid-Schiff (PAS) stain (Fig. 1C), which strongly suggested Lafora bodies.
Third-generation sequencing of the proband (III 3) revealed two mutations, c.476+14860 C > A and c.301+1 G > A in EPM2A (NM_005670.4, GRCh37/hg19). Pedigree verification showed that the patient’s mother carried c.476+14860 C > A and father carried c.301+1 G > A. The compound heterozygous mutations were also identified in the proband’s younger brother (III 4) and cousin (III 8) (Fig. 1A). Sanger sequencing further confirmed the deep intronic mutation c.476+14860 C > A caused aberrant splicing (Fig. 1D). We further verified aberrant splicing by reverse transcription PCR (RT-PCR). The results of RT-PCR revealed that, other than the normal product, shorter-than-expected sequence were found in patient using primers spanning c.476+14860 C > A (Fig. 1E). Western blot showed no EPM2A protein in the muscle tissue of patient (Fig. 1F).
Nanopore RNA sequencing was carried out using proband’s muscle tissue. A zoom-in view showed that EPM2A c.301+1 G > A caused aberrant splicing at donor site and resulted in intron retention (referred to NM_005670.4) according to GU-AG rule (Fig. 2A, C). EPM2A c.476+14860 C > A would cause would cause an extra splicing and protein truncation (Fig. 2B). Sashimi plot analysis of the RNA-Seq data showed extra “ribs” around c.301+1 G > A (Fig. 2C, indicated by blue arrow), suggesting the retention of intron. We also observed additional connecting lines between exons near c.476+14860 C > A (Fig. 2C, indicated by green arrow). The variant c.476+14860 C > A lies in the deep intron according to transcript variant 1 (NM_005670.4) but locates in exon in transcript variant 6 and 10 (NM_001368129.2 and NM_001368132.1). This variant follows closely behind GC dinucleotides which caused formation of a novel splicing donor based on GC-AG rule and resulted in exon truncation (Fig. 2D).
Fig. 2
Integrative Genomics Viewer and schematic diagram of the biallelic intronic EPM2A mutations. A Integrative Genomics Viewer (IGV) showing intron retention caused by EPM2A c.301+1 G > A in the proband and normal splicing in healthy controls. B IGV showing exon truncation caused by EPM2A c.476+14860 C > A and normal transcripts in healthy controls. C Sashimi plot analysis suggests the intron retention and extra splicing in the patient. The number of reads supporting the prediction of splicing is marked on the connecting lines. D Schematic diagram of the abnormal splicing caused by EPM2A c.301+1 G > A and c.476+14860 C > A. The transcripts are numbered according to https://www.ncbi.nlm.nih.gov/gene/
留言 (0)