
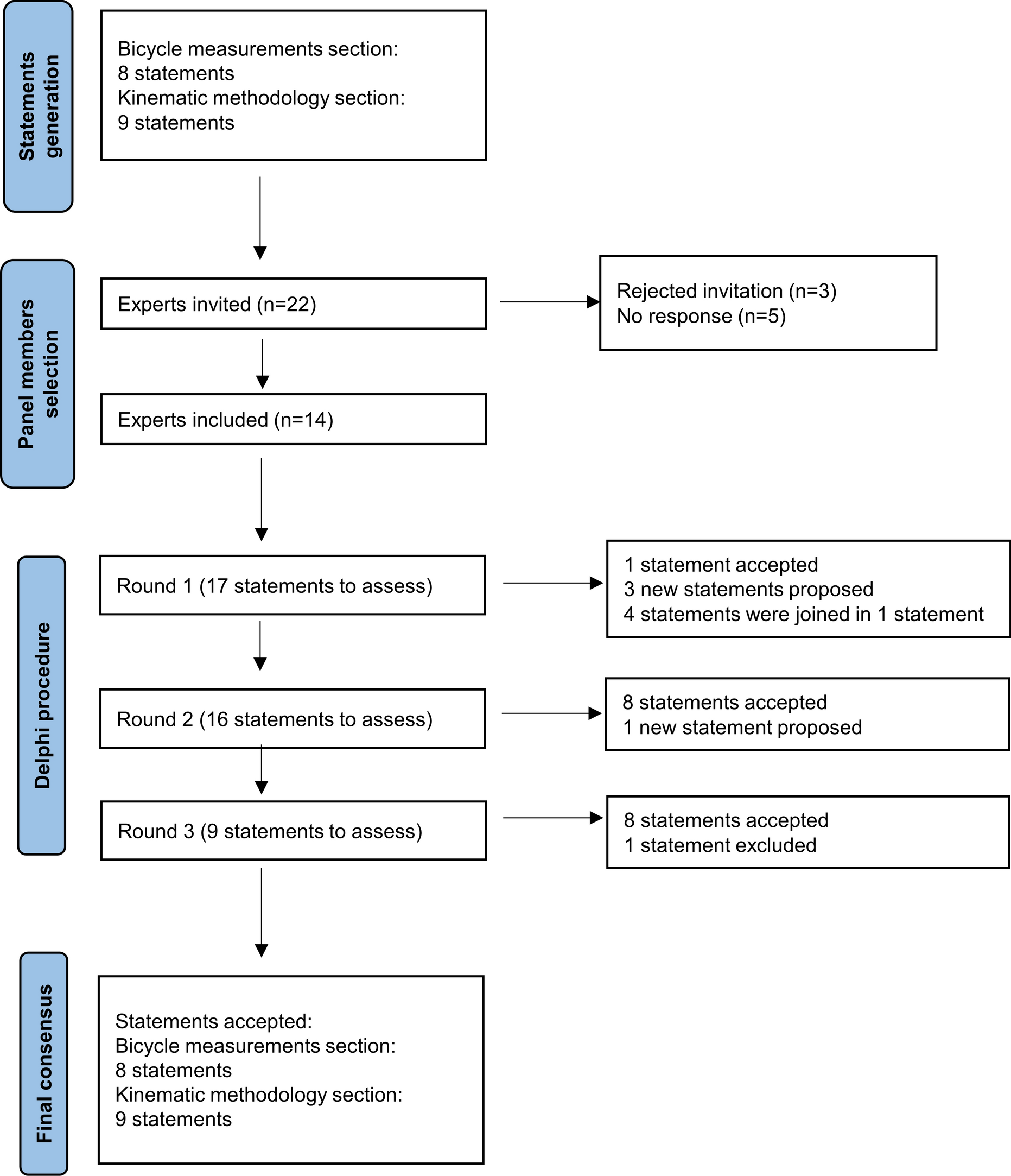
記住我
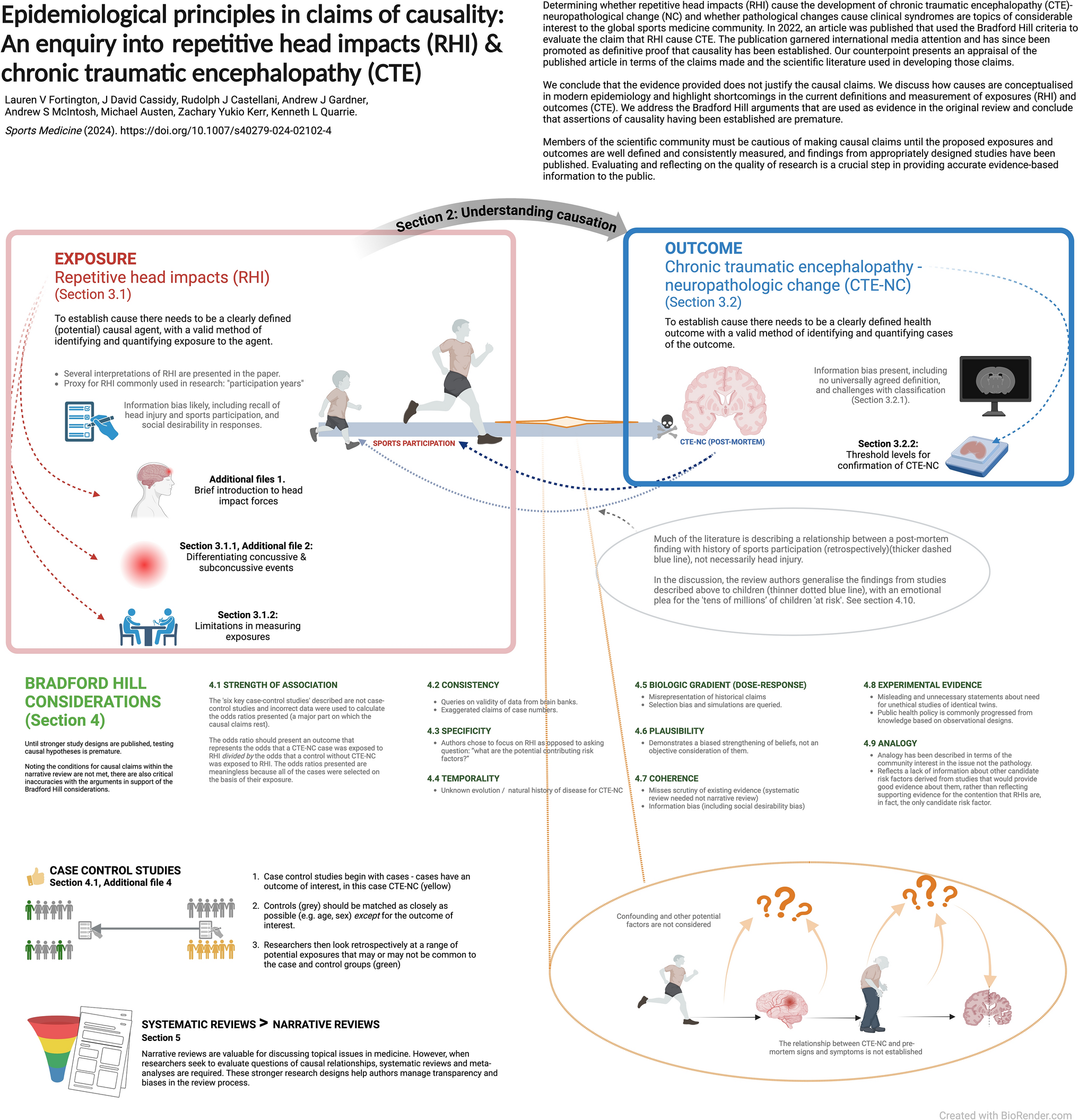

Bradford Hill’s considerations were developed under the assumption that there are results available from hypothesis-testing studies, such as case–control and cohort studies. To the best of our knowledge, findings of studies using such designs to examine whether RHIs cause CTE-NC, and whether CTE-NC represents a progressive neurodegenerative disease, have not yet been published.
Beyond the fact that the conditions for causal claims are yet to be met, we believe the statements by the authors [1] implying that the application of the Bradford Hill considerations means that RHI has been established as the cause of CTE contains several significant inaccuracies. In the sections below we provide our reasoning for our belief, and highlight research methods and approaches to public health and risk management that we think can better address the concerns raised by the authors [1].
4.1 Strength of AssociationThe strength of association among variables is often presented via statistics such as correlations, risk and rate differences and hazard, risk, rate and odds ratios. While there is a general premise that a stronger (positive) association between an exposure and outcome is more likely to be a relationship that is causal in nature, this does not always hold:
…a strong association is neither necessary nor sufficient for causality… [62]
The review authors describe ‘six well-conducted case–control studies where the researchers made a reasonable attempt to identify RHI history and had more than 50 subjects to be sufficiently powered for statistical significance…’ [1, p 4]. The data available in the six studies provide either weak evidence, due to a mix of ascertainment bias and unclear validity of proxy measures used to estimate exposure to RHI, or are unsuitable to report odds ratios reflective of the likelihood of developing CTE-NC on the basis of exposure to RHI due to the way cases and controls were selected. We agree with the authors of a systematic review that these six studies are not case–control or cohort designs [50].
Because these studies do not meet the requirements of a case–control design, the odds ratios presented are invalid. As an example, one of the key sources is the VA–BU–CLF brain bank, which sought inclusion of cases on the basis of their exposure to RHI in the first place.
All 269 brains from the VA–BU–CLF Brain Bank had contact-sports history. [1]
If cases are selected into a case–control study because they were exposed, the odds of exposure in the case group are meaningless. In turn, the odds ratio comparing exposure in the case group with that of the controls does not represent the relationship between development of the outcome and exposure to the agent. The selection of cases on the basis of exposure is why some of the calculations reported required imputed values, and why the odds ratios appear unusually large. In addition, because the prevalence of CTE-NC is so high in the VA–BU–CLF brain bank, applying the rare disease assumption to the use of odds ratios to approximate relative risks is inappropriate, and yields a gross overestimate of the risks [63]. Refer to Additional files 4: Case control studies and odds ratios.
4.2 ConsistencyConsistency refers to repeated findings within and between studies, settings, timepoints and populations, and similar to strength of association, consistency does not necessarily imply a causal relationship, nor does lack of consistency rule out a causal association [62]. In their Table 3, the review authors present ‘CTE cases diagnosed globally’ as justification for causality through consistency, with a summary of case series published by different brain banks/groups [1]. The consistency of brain bank data demonstrating both CTE pathology together with a retrospective history of RHI, regardless of whether co-morbid pathology is present, is difficult to accept as evidence for causation, given the sources of error with outcomes and exposures already described.
‘McKee et al. (reference number 18 [64],) noted that prior to 2009, there were only 48 cases of CTE in the literature, in contrast to the hundreds of cases of CTE since (reference numbers 19–30)’ [1, p 2]. Further, in their Table 3, the authors present ‘the largest CTE case series’ published at various brain banks that are ‘understood to be using NINDS/NIBIB consensus criteria for diagnosis’ [1, p 5–6].
These data (from references 19–30 and Table 3) on the number of CTE cases diagnosed globally are presented as justification for causality through consistency. There are overlapping cases from the VA–BU–CLF brain bank (reference numbers 19, 20, 21, 22) and we are unable to differentiate the exact number of unique cases from these series. Table 1 summarises the sources cited, noting critically that not all cases in this table are confirmed with histopathological CTE-NC. Some findings were based on the first NINDS/NIBIB consensus meeting (published in 2016), some cases have multiple diagnoses, and two studies were published in 2015, before the first NINDS/NIBIB criteria were reported.
We emphasise that generalisations and exaggeration are not helpful for understanding the natural history or pathology of CTE-NC and any potential relationship with RHI. The information further empasises the importance of defining and measureing exposures and outcomes accurately.
4.3 SpecificityRHI is the only factor common to reported CTE cases, and there is almost no evidence of CTE in those examined who have not sustained RHI [1, p 7].
We believe the claims made by the review authors [1] in support of this section to be misleading of the current literature, particularly as they chose to focus on RHI as the lead risk factor for CTE-NC, as opposed to exploring the question ‘what are the potential contributing risk factors?’.
There are several alternative, and reasonable, factors explored in the literature that may cause CTE-NC, either independently of, or in conjunction with, RHI, including genetics, inflammatory responses, ergogenic aids and substance use to name a few [79, 80]. There is also more than ‘almost no evidence’ of CTE-NC without RHI, with various cases from literature having been described [81, 82]. Finally, irrespective of the issues identified, and sufficient alone, is that the absence of evidence is not evidence for its absence. Rather, we need to continue asking the right questions and addressing them with suitable study designs.
4.4 TemporalityTemporality is perhaps one of the more intuitive concepts to understand in establishing cause: the risk factor (or exposure) must occur before the disease (or outcome). While temporality seems straightforward, until there are clear parameters to define and measure RHI and better information regarding any clinical manifestation of CTE-NC is available, this viewpoint also remains uncertain.
Establishing temporal associations of RHI and CTE-NC is challenged by not knowing the evolution of CTE-NC before, during and after RHI exposure, whether any pathology becomes stationary, whether it is reversible, or whether it is progressive (and why that might be the case in the absence of further exposure to RHI) [83]. The authors write:
As outlined in the above section on specificity, the exposure to RHI is associated with CTE pathology and, especially with the introduction of the aforementioned revised NINDS/NIBIB neuropathologic criteria requiring neuronal involvement in the perivascular deposition of tau, this pathology occurs nearly exclusively in the presence of clearly identified RHI exposure [1, p 7].
Here, the authors have exemplified several of our concerns with the claim of ‘RHI’ being a ‘clearly identified exposure’, as presented earlier.
4.5 Biologic Gradient (dose–response)This section of the causal claims rests partly on a description of historical cases. The condition termed ‘CTE’ in reports of ‘dementia pugilistica’ among boxers is qualitatively different from that which is currently termed CTE-NC. This is incorrect and misleading of what is currently known about CTE-NC.
The following two quotes exemplify how the authors of the review equate ‘dementia puglistica’, or ‘punch drunk’ syndrome, with modern conceptions of CTE:
Dr. Harrison Martland is credited with first identifying the syndrome that was later called CTE in his article Punch Drunk, published in the Journal of the American Medical Association in 1928 [1, p 7].
While CTE has been known in the literature for nearly a century, most of the research on CTE has occurred only in the last decade [1, p 2].
These statements leave the reader with an impression that historical punch-drunk syndrome and modern day CTE are exchangeable when, in fact, they are markedly different. Punch-drunk syndrome, characterised by clinical signs such as dysarthria, shuffling gait and Parkinson’s-type symptoms, was identified and conceptualised on the basis of clinical neurological examination (multiple and variable neurological deficits from extreme neurotrauma exposure). Modern CTE-NC is a purely neuropathological finding (or more specifically, an immunohistochemical finding) that, thus far, lacks a specific clinical presentation. Neither Martland nor Critchley reported pathological changes as is implied. Further, the work of Goldfinger et al., who re-examined the Corsellis series using the 2016 NINDS/NIBIB criteria and modern immunohistochemical techniques, refuting several of the original Corsellis findings, has been overlooked in the narrative review [84].
Focal deficits attributed to boxing such as slurring dysarthria, tremor and gait disturbances at or before retirement, were common in early twentieth-century boxers with prolonged neurotrauma exposure. These visible signs are not commonly seen in the case of modern athletes; for example, one is hard pressed to find even a single case of an American football player with a focal neurological deficit at retirement. Further information is presented in Additional files 3—Misrepresentations of historical research.
The issue of selection bias is also raised in this section on biological gradient. Selection bias describes a systematic difference in the relationship of exposure and disease between those who participate in a study and those who in theory could be eligible for the study but did not partake [85]. In the case of brain bank cohorts, participants are not randomly assigned to be investigated, rather they or their next of kin choose to donate their brain, often because of specific health concerns. Because the donation of brains into many brain banks is based on symptoms during life, as well as contact sport or RHI exposure, apparent relationships found in the data may not generalise to the wider populations.
To account for selection bias, the authors [1] refer to the findings of LeClair et al. [86], who explored the influence of selection bias through simulated analyses. While the use of quantitative methods of bias analysis is endorsed by experts in epidemiological statistics, such as Greenland [87] and Lash et al. [88], both highlight that the methods often require the use of unverifiable assumptions about probabilities of selection and non-selection across groups. To the extent that modelling does incorporate such assumptions, the results of sensitivity analyses reflect plausible conjectures about the effects that would have been found had selection bias not been a feature of the study, rather than direct evidence of the size and direction of the true effect. As Lash notes, ‘…bias analyses do not establish the existence or absence of causal effects any more than do conventional analyses’ and ‘…when examining a bias analysis, a reader must bear in mind that other reasonable inputs might produce quite different results’ [88, p 714].
In relation to the LeClair study, Nowsinki et al. write ‘the researchers found that highest level of football play was associated with CTE diagnosis in a dose–response manner’ [1, p 9]. However, as described in the study limitations by LeClair et al., ‘exposure’ was treated as a categorical variable in which the ‘…highest level of American football playing served as a proxy measure for RHI…’ and ‘we were unable to consider other measures of exposure, such as frequency of RHI, or even duration of play…’ [86, p 1441]. Ultimately, the ‘dose’ argument of RHI has shifted well away from being ‘the cumulative exposure to recurrent concussive or subconcussive events’ [1, p 2].
4.6 PlausibilityFindings from animal and simulation studies are provided in the narrative review as examples that ‘provide evidence of a credible mechanistic hypothesis for the location of the pathognomonic lesion and the association between RHI and CTE alongside the paucity of CTE cases in individuals not exposed to RHI all support that RHI exposure is a plausible cause of CTE’ [1, p 9]. We accept that the evidence presented in this section is consistent with the hypothesis that RHIs may be a causal factor in CTE-NC. Further, we recognise the value that animal studies and simulations have in understanding the aetiology of human disease processes, challenges in translating findings from animals to humans notwithstanding. As Shimonovich et al. [24] write, however: ‘the plausibility of the causal relationship is both dependent on and limited by knowledge available at the time. It may be further limited by assumptions based on investigators’ beliefs rather than empirical evidence’ [24, p 882].
4.7 CoherenceCoherence requires that what is known about the cause and effect proposed does not conflict with what is known about the natural history of disease. The review authors state ‘we must demonstrate that the association between RHI and CTE pathology does not conflict with what we know about the development of CTE pathology or RHI’ [1, p 9]. As pointed out elsewhere, however, coherence, ‘provides at best only weak support for causality, because many theories will exhibit such coherence, including most theories that are proposed and eventually refuted’ [89, p 21].
In their section on coherence, the authors [1] briefly consider the potential of other causal variables. The authors focus on opiate misuse as the only variable that ‘has been proposed as a potential alternative cause of CTE’, subsequently dismissing it on the basis of one study that reported ‘tau deposition from opiate use is easily distinguished from the pathognomonic CTE lesion’. They conclude the section with the following statement: ‘With what is known in the literature about computer modeling of brain trauma, post-mortem confirmed cases without a history of RHI exposure, sex differences, opiate use, and CTE genetics, RHI remains the only candidate risk factor for CTE causation’ [1, p 10].
We recommend further scrutiny of existing evidence before drawing conclusions from these arguments of coherence, not least because studies that would permit proper evaluation of a range of possible contributing factors to the development of CTE have not yet been conducted. We note that other authors have raised multiple candidate risk factors for both CTE-NC and clinical and functional outcomes, including pre-existing psychiatric conditions, sleep disorders, substance use, chronic pain, genetic factors and exposure to anaesthesia [79, 90]. We do not believe that the statement that RHI is the only candidate risk factor for CTE causation is well supported by the evidence at this point.
4.8 Experimental EvidenceAlthough all study designs used in epidemiology have their limitations [87], findings from well-designed cohort studies and case–control studies are generally accepted among epidemiologists as capable of testing causal hypotheses. Randomised controlled trials are best suited to testing causal hypotheses but are limited in their application for many public health issues, including injury. Clinical case-series and cross-sectional studies have important roles in epidemiology, especially with respect to identifying novel health outcomes and developing hypotheses to be tested in more rigorous designs. Case-series and cross-sectional studies also have significant, and well-recognised, limitations with respect to generalising results from the individuals and groups studied to the wider population.
Establishing causation (or not, as the case may be) between RHI and CTE-NC can in principle be ascertained via the application of observational research designs [91]. Such studies need to be designed to properly account for the effects of random error, confounding, information bias and selection bias. Prospective and retrospective cohort studies, as well as case–control studies, could be developed that would provide answers to many important questions including:
whether, and to what extent, RHIs are a causal factor of CTE-NC;
whether, and to what extent, factors other than RHIs cause CTE-NC;
whether CTE-NC represents a progressive neurological disease; and
whether, and to what extent, CTE-NC causes the range of clinical outcomes to which it has been linked via cross-sectional analyses.
The same or similar studies [92] could simultaneously address the effects of RHI on other health outcomes of interest, such as depression, neurodegenerative diseases and dementia, and whether they were related to CTE-NC.
Once definitions of RHIs and CTE-NC are developed, agreed on and validated, cohort (retrospective and prospective) and case–control studies are likely to provide much stronger evidence of the relationship between RHIs and CTE-NC than has yet been presented, at which point cautious judgements about the likelihood of observed relationships being causal can, and should, be made. In their discussion section, the authors have implied such studies are impossible to conduct, with the claim that they would require unfeasible studies of identical twins and unethical assignment to groups which are, and are not, subjected to head injuries from early in life. We disagree with that view of study design and reiterate that much research in public health is conducted using observational designs, with true experimental designs unsuitable in many public health scenarios [93].
4.9 AnalogyIn this section, analogies are drawn between the level of evidence that was obtained regarding the causal relationship between cigarette smoking and lung cancer being ‘well established’ with that between RHI and CTE-NC, and further between issues regarding how exposure to cigarette smoke has been quantified in observational studies and how exposure to RHIs have been quantified.
In their review [1, p 11], the authors claim that with respect to exposure to cigarette smoking ‘key questions remain unanswered or incompletely answered, including what precisely constitutes a smoked cigarette (the dose), why some smokers develop cancer and others do not, how many cigarettes are too many, or which specific cigarette or carcinogen sparked the lung cancer’ [1, p 11]. No cited evidence is provided in support of their claim that the lack of a precise measurement of a smoked cigarette is actually a feature of the epidemiological evidence, but they do use it to set up the following argument: ‘The fact that these questions also remain for RHI and subconcussive impacts is often raised as a reason that conclusions on RHI/CTE causation cannot be drawn’ [1, p 11]. They conclude that: ‘These knowledge gaps have not limited the ability to assert a causal link between smoking and lung cancer, and similarly should not limit the ability to determine the likelihood of a causal link between RHI and CTE’ [1, p 12].
The argument is an example of the ‘straw man’ fallacy. Focussing on the first claim, regarding exposure to smoked cigarettes, a systematic review and meta-analysis examining survey-based assessments of exposure to cigarette smoking published in 1994 found that self-reported smoking status had generally high levels of sensitivity (87%) and specificity (89%) when validated against biochemical measures of exposure across the 26 studies [94]. Although it is acknowledged that survey methods provide less accurate information in some situations (for example, when assaying cigarette use among pregnant women), [94] there is no doubt in the epidemiological community that measures of ‘dose’ captured through surveys asking about cigarettes smoked per day or pack years of exposure have yielded valid information regarding the link between smoking and lung cancer.
The qualitative and quantitative differences in the amount of evidence regarding smoking causing lung cancer and RHIs causing CTE-NC are currently so large that claims of the two issues being comparable are misleading. As noted above, the studies identified in the review [1] regarding the relationship between RHI and CTE-NC have employed a range of approaches that have yet to be validated in assessing exposure to RHIs, along with definitions of CTE-NC that have varied over time. To date, studies have primarily used case-series and cross-sectional designs with papers from overlapping subsets of the VA–BU–CLF brain bank case series providing the data for the great majority of the existing publications, as well as the case material for consensus efforts. The autopsy case-series data have been supplemented by interviews and surveys of ‘informants’, who are predominantly next of kin of the deceased.
By contrast, the use of case–control and cohort designs using consistent methods of appraising exposure are a feature of the studies examining the relationship between smoking and lung cancer in humans. The amount of supporting evidence for the contention that cigarette smoking causes lung cancer differs from that regarding RHIs and CTE-NC by an order of magnitude. A systematic review and meta-analysis published in 2012 of the results of studies published up till the year 2000 identified 267 ‘principal’ (and 20 subsidiary) studies, of which 209 used case–control and 52 used prospective cohort designs [95].
Questions such as ‘which specific cigarette sparked the lung cancer’ or ‘which specific head impact sparked the development of CTE-NC’ might form the basis of legal arguments or judgements regarding insurance claims but are not questions that epidemiological studies would address because the questions are directed at individuals, not at population groups. The question of why some smokers develop lung cancer while others do not is relevant to considerations of how cause is conceptualised, but as discussed by Brand and Finkel [96] and reiterated in the review [1], the fact that a cause of a health outcome is a cause does not necessarily imply that all those exposed to it will develop the outcome, nor that people not exposed to it will not. It does, however, imply that factors other than the specified cause contribute to the outcome.
Cohort study designs, which permit the evaluation of multiple potential mediators and confounding variables are valuable in enabling understanding of how strongly associated with an outcome a given cause is, and how that cause interacts with other potential causes and confounding factors. The claim that RHIs are ‘the only candidate risk factor for CTE causation’ [1, p 10] reflects a lack of information about other candidate risk factors derived from studies that would provide good evidence about them, rather than reflecting supporting evidence for the contention that RHIs are, in fact, the only candidate risk factor.
4.10 Comments on Children’s Participation in SportIn the discussion section of the review, the authors emphasise the question: what impact do head injuries sustained in youth sport have on participants in the long term? writing:
Perhaps most consequential would be the positive health impact for children. As it stands today, tens of millions of children as young as 5 years old are exposed to RHI in sports because they are playing by rules that were originally designed for adults. Armed with confidence in the causal connection between RHI and CTE, parents and youth coaches may reject exposing their children to a preventable degenerative brain disease simply because the current rules (tackling, heading) make RHI inevitable, especially when non-RHI versions of those contact sports exist, as well as alternative sports without RHI. Considering that both CTE onset and severity have been associated with a dose-response, strict reforms that lower the dose could effectively prevent new cases of the disease [1, p 13–14].
This addition to the paper doesn’t follow from the prior content, because evidence about the effects of children’s sport on health outcomes was not presented. Rather, it serves as an appeal to inherent emotional concerns. The statement was made upfront that ‘any reference to CTE in this review refers to cases that have been confirmed by autopsy’ [1, p 3] so declarations that ‘parents and youth coaches may reject exposing their children to a preventable degenerative brain disease’ [1, p 14] is overreaching.
In a 2019 narrative review into the age of first exposure to tackle football and later life outcomes Alosco and Stern stated ‘…it is our opinion that more methodologically rigorous research on the long-term neurologic consequences of youth tackle football is needed before policy and safety guidelines can be accurately informed’ [97, p 113]. Further, in a 2021 narrative review, Iverson et al. [98, p 1] concluded ‘The accumulated research to date suggests that earlier AFE (age of first exposure) to contact/collision sports is not associated with worse cognitive functioning or mental health in (i) current high school athletes, (ii) current collegiate athletes, or (iii) middle-aged men who played high school football. The literature on former NFL players is mixed and does not, at present, clearly support the theory that exposure to tackle football before age 12 is associated with later in life cognitive impairment or mental health problems’. More recently, in 2023, the Concussion in Sport Group did not find evidence to support the notion that participants in youth, high school and collegiate sports are at risk for long-term consequences [50].
Of course, encouraging individuals and sports organisations to reduce exposure to brain injuries is sensible, and sports organisations have made relevant changes that are being closely monitored. For example, the age for body-checking in youth ice hockey was raised [99,100,101,102]. There has also been a general cultural shift to understand reasoning for, and discourage, poor compliance from players, parents or others with changes that are implemented to reduce exposure to, and consequences from, brain injuries sustained in sports [
留言 (0)