
記住我






Anticancer treatment consists of an armamentarium of many modalities, like surgery, chemotherapy (CT), radiotherapy (RT) and immunotherapy (IT). In the last decade, treatment with ITs has emerged as an extremely power tool for the treatment of cancer. Immunotherapy can deliver personalised treatment as per the oncogenic profile of a specific target in various solid malignancies. The human immune system can adapt dynamically to keep pace with the rate of mutation and growth of cancer cells (Tang et al., 2014). The immune system has an innate ability to “remember” cancer cells; therefore IT, can offer targeted treatment and protection against cancer recurrence. The majority of targeted drugs exhibit restricted effectiveness against solid tumors, primarily attributed to the frequent development of resistance to these treatments (Vaddepally et al., 2020). In recent times, IT such as adoptive cell treatment and immunological checkpoint blockade (ICB), has demonstrated impressive clinical effectiveness. The use of ICBs to treat solid tumors has been authorised for cancer treatment targeting a wide range of molecules, including CTLA4, PD1, and ligand 1 (PDL1) (Zhu et al., 2021a). Anti-PD1 therapy has emerged as a leading form of ICB therapy, outperforming the anti-CTLA4 therapy in various tumor types. However, its efficacy as a stand-alone treatment is typically limited, with a response rate of only about 20% particularly for the advanced stage cancers (Kim et al., 2022). In addition, cancer cells often develop adaptive immune resistance mechanisms to evade immune system attacks. Given these challenges, combining IT with complementary approaches is a rational strategy to enhance the anti-tumor effects (Zimmer et al., 2020; Zhu et al., 2021b).
A key component of cancer care is RT in combination with surgery and systemic therapies such as IT, CT, and targeted therapies. The primary goal of RT is to enhance the delivery of radiation to the tumor, thereby ensuring local control while minimising radiation exposure to the adjacent healthy tissue. Advancements like High Linear Energy Transfer (LET) or Intensity-Modulated RT (IMRT) have significantly improved the therapeutic ratio (Elshaikh et al., 2006). IMRT indeed represents a significant advancement in RT techniques. Its ability to modulate the intensity of radiation beams allows for more precise targeting of tumors while sparing the surrounding healthy tissues. This precision translates into better tumor coverage and reduced toxicity compared to the conventional RT. IMRT achieves this by dividing the radiation beam into many smaller beamlets, each with varying intensity levels. By adjusting the intensity of these beamlets and their angles, radiologists can tailor the radiation dose to conform closely to the tumor area, delivering higher doses to the tumor while minimising the radiation exposure to the nearby normal tissues (Eisbruch et al., 1998). However, despite these significant advances, many patients still encounter local recurrences of the disease following RT. Most of the DNA damage, particularly the double-strand breaks (DSBs), is what causes the RT-induced cell death. As a result, tumor cells with effective DNA repair systems are resistant to ionizing radiation, but tumor cells with impaired DSB repair pathways are more susceptible to death (Zhu et al., 2021a). Therefore, treatments of the tumor cells with small molecule inhibitors that block or impair the machinery responsible for repairing DNA damage may improve the overall effectiveness of RT.
CT, another prevalent form of genotoxic treatment for cancer therapy, employs a class of pharmaceutical small molecule agents that induce DNA damage through various mechanisms, including topoisomerase inhibition, DNA alkylation, and crosslinking (Elshaikh et al., 2006). Often administered alongside RT or surgery, CT has been demonstrated to influence the host immune response. Both CT and RT have been shown to interact with the immune checkpoint inhibitors (ICIs) in a synergistic manner.
The DNA damage response (DDR) plays a crucial role in preserving the genomic stability through the restoration of various forms of DNA damage in cells (Jeggo and Lavin, 2009). Compared to normal cells, cancer cells with high underlying levels of DNA damage and genomic instability depend more on DDR for the survival (DNA damage accumulates because of DDR deficiencies, which also increase tumor immunogenicity). Malfunctions of the DDR pathway can arise from mutation(s) in various genes involved in DDR pathway and/or due to epigenetic modifications of the DDR proteins that result in the impaired and/or dysfunctional DDR (Chabner and Roberts, 2005). Therefore, combining CT, IR therapy, and IT with DDR network inhibitors has been drawing increasing attention in a large number of clinical trials.
In this review, we will explore various aspects, including different types of DNA damage and their repair pathways, the synergy of DDR with various cancer treatment options such as RT, CT, and IT.
DNA damage response in eukaryotesAll organisms must maintain the DNA sequence integrity and fidelity of their genome to survive and function properly. The eukaryotic genome faces continuous challenges from a wide variety of external and internal sources of DNA damage, including reactive oxygen species (ROS), IR, UV light and various exogenous chemical agents that can induce different types of damages in genome (Das et al., 2023). To address the fundamental issue of genomic erosion, organisms have developed an intricate network of the DDR systems. This network encompasses various damage tolerance processes, DNA repair mechanisms, and cell-cycle checkpoint pathways. The primary regulators of DDR signalling pathway that are activated via phosphorylation in response to DNA damage is the Ataxia-telangiectasia mutated (ATM) and Ataxia-telangiectasia and Rad3 related (ATR) kinases. ATM, a serine-threonine kinase serves as the primary orchestrator in the cellular response to DNA DSBs induced by exposure to IR and stalled replication forks. On the other hand, ATR plays a major role in single-strand DNA damage repair mechanism (Jurkovicova et al., 2022). Two extensively studied downstream targets of ATM and ATR are the cell cycle checkpoint kinases CHK1 and CHK2. The coordinated action of these proteins initiate the ATM and/or ATR kinase-initiated signalling cascade and play critical roles in determining the cellular responses to DNA damage (Dagar et al., 2023). The activated CHK1/CHK2 kinases further phosphorylate p53 and CDC25. The activation of this phosphorylation cascade results in increased expression of p21 (regulated by p53), and inhibition of CDK activity, which ultimately leads to cell cycle arrest at the G1-S and G2-M transitions (regulated by CDC25 and WEE1) (Mushtaq et al., 2021).
The molecular pathways of DNA repair mechanisms involved in addressing common types of DNA damage are the nucleotide excision repair (NER), homologous recombination (HR), non-homologous end joining (NHEJ), base excision repair (BER), and mismatch repair (MMR). These DNA damage repair pathways play crucial roles in maintaining the genome integrity of mammalian cells.
Moreover simple solution that has evolved in response to DNA damage involves the direct reversal of the DNA lesions through the activities of specialized enzymes. Examples include photolyases, which selectively reverse the UV-induced DNA damage (Jurkovicova et al., 2022), and the suicide enzyme O6-methylguanine transferase (MGMT), which is involved in repairing specific types of DNA lesions. It operates by repairing damaged guanine nucleotides and transferring the methyl group at the guanine’s O6 site to its cysteine residues. This enzymatic activity helps prevent gene mutations, cell death, and the onset of tumorigenesis induced by alkylating agents (Groelly et al., 2023). Because the photolyases are not conserved in mammals, they heavily rely on intricate molecular processes for the repair and elimination of the UV-induced damages viz. NER.
Nucleotide excision repair eliminates a diverse range of single-strand lesions that cause local helix destabilization. NER is a multifaceted, intricate multistep process that requires the coordinated action of around 25 distinct polypeptides and plays a pivotal role in eliminating bulky DNA intra-strand and interstrand crosslinks (ICL) adducts (Weber, 2005). NER plays a pivotal role in eliminating bulky DNA adducts on DNA and contributes to the repair of intra-strand and inter-strand crosslinks (ICLs). The xeroderma pigmentosum (XP) proteins, along with excision repair cross-complementation group 1 (ERCC1), both play essential roles in the NER pathway. NER is involved in two types of repairs: global genome NER (GG-NER) and transcription-coupled NER (TC-NER). If the damage occurs within the actively transcribed strands of genes, TC-NER mechanism is activated. In the TC-NER mechanism, the detection of DNA damage in the transcribing DNA is carried out by the stalled RNA polymerase. The Cockayne syndrome factors A and B (CSA and CSB) play essential roles in orchestrating the formation and function of the TC-NER complex (NCBI, 2024b). If damage is not in the actively transcribed strand of a gene, then GG-NER is initiated. A dimer consisting of XPC and HR23B appears to recognize and bind to the damaged DNA. This is followed by the binding of the general transcription factor TFIIH and XPA, a DNA binding protein. The binding of XPA facilitates the binding of Replication Protein A (RPA). This heterotrimer complex stabilizes the unwound DNA and guides the two structure-specific endonucleases, the ERCC1-XPF complex and XGP. Excision of damaged DNA is followed by the replicative gap-repair proteins that carry out DNA synthesis, the final nick is sealed by DNA ligase I (Hoeijmakers, 1993).
Lesions that serve as substrates for both NER and BER are situated within one of the DNA strands. Some genotoxic agents, such as IR, and various chemotherapeutic drugs, affect both the strands of DNA and induce DSBs which are repaired by two distinctly different pathways: NHEJ and HR (Groelly et al., 2023). HR is characterized by its high fidelity of DNA repair. The process initiates with the recognition and processing of the damaged DNA by the CtlP and MRN (MRE11-RAD50-NBS1) complex through end resection, enabling the binding of RPA to the single-stranded overhang. The PALB2, BRCA1, and BRCA2, and complex recruit RAD51, which displaces RPA. Through strand invasion, the processed DNA filament attaches to the intact DNA to create the new complementary DNA strand (Fousteri and Mullenders, 2008).
The NHEJ mechanism stands out from the HR as it does not rely on template DNA for the repair process as NHEJ is an error-prone repair mechanism. NHEJ breaks are recognized by Ku heterodimer (Ku70/80) subunits that activate DNA-PK, a PI3-kinase. XRCC4 and DNA ligase IV are also recruited to the DSB sites to ligate the DNA ends and fill in any gaps in the sequence. Later, XRCC4, Pol μ, and DNA Ligase four are then recruited to the DSB sites and ligate the DNA ends together (Giglia-Mari et al., 2011). In the absence of essential NHEJ components, the Alternative End Joining (A-EJ) pathway, also referred to as microhomology-mediated end joining, becomes more prominent in response to DDR. A-EJ relies on PARP1 and DNA polymerase theta (Pol θ), to facilitate the re-joining of two DNA ends, utilizing very short homologous sequences typically ranging from 2 to 20 base pairs (San Filippo et al., 2008). Decreased expression or loss of certain NHEJ proteins, such as Rev7 and 53BP1, can result in resistance to PARP inhibitors, particularly in BRCA1-deficient cancers.
The BER pathway corrects minor base lesions that do not significantly disrupt the DNA double-helix structure. The key elements of the repair pathway include DNA polymerases, endonucleases, glycosylases, and DNA ligases. PARP1 and PARP2 help to facilitate the process (Burma et al., 2006). Damage bases are first identified and removed by DNA glycosylases creating apurinic or apyrimidinic (AP) sites. Both PARP1 and apurinic/apyrimidinic endonuclease 1 (APE1) can detect and bind the damage sites. This triggers the catalysis of poly ADP-ribosylation (PAR) on various protein substrates, facilitating the recruitment of repair proteins to the site of damage. The subsequent synthesis and ligation step of BER is bifurcated into two sub-pathways—short-patch and long-patch (Ceccaldi et al., 2015). In short-patch BER, the gap is filled with the correct base pair by polymerase beta (Pol β). The consecutive ligation of DNA ends requires either the DNA ligase I (LIG1) or the complex formed by X-ray repair cross-complementing protein 1 (XRCC1) and DNA ligase III (LIG3). While in long-patch BER, the process involves lap endonuclease-1 (FEN1), proliferating cell nuclear antigen (PCNA) (NCBI, 2024c) polymerase delta/epsilon (Pol δ/ε), replication factor-C (RFC), and LIG1 (Barakat et al., 2012).
Preserving the genome stability is largely dependent on MMR system, a biological pathway that is highly conserved. Base-base mismatches and insertion/deletion mismatches produced during DNA replication and recombination are the main sources of MMR’s specificity (Maynard et al., 2009). Additionally, MMR inhibits HR and has been implicated in the signalling of DNA damage in eukaryotic cells. An MSH/MSH6 (MutSa) or MSH2/MSH3 (MutSb) ATPase heterodimer is assembled to produce a sliding clamp that is responsible for detecting and initiating the DNA repair (Li, 2008). Other elements are brought to the site after the mismatch is identified. With endonuclease activity, the MLH1/PMS2 (MutLa) complex makes a first cut in the helix, which is followed by a larger EXO1-mediated resection. MSH2-MSH3 detects longer lengths of mismatches, while the MSH2-MSH6 complex detects shorter ones. MLH1- PMS2, PMS2, and EXO1 are involved in excising the mismatched DNA (Strzalka and Ziemienowicz, 2011).
IR-induced DNA damage in cancer therapyIR is a cornerstone in cancer therapy, leveraging its ability to induce DNA damage to eradicate malignant cells. Therefore, understanding the biochemical and molecular basis by which IR induce DNA damage can provide useful information. Understanding these mechanisms is crucial for comprehending the genotoxic effects of IR. There are some primary mechanisms of IR-induced DNA damage, such as direct ionization which directly interacts with the DNA molecule, leading to ionization. It causes the ejection of electrons from atoms within the DNA molecule, resulting in the formation of ion pairs and free radicals, or an indirect effect through water radiolysis. In this mechanism, IR, indirectly induce DNA damage by ionizing water molecules in the cellular environment through a process known as radiolysis (Fortini and Dogliotti, 2007). Radiolysis of water produces ROS, such as hydroxyl radicals (•OH), hydrogen peroxide (H2O2), and superoxide radicals (O2•−). These ROS can then interact with DNA and cause damage (Barakat et al., 2012) (Figure 1).
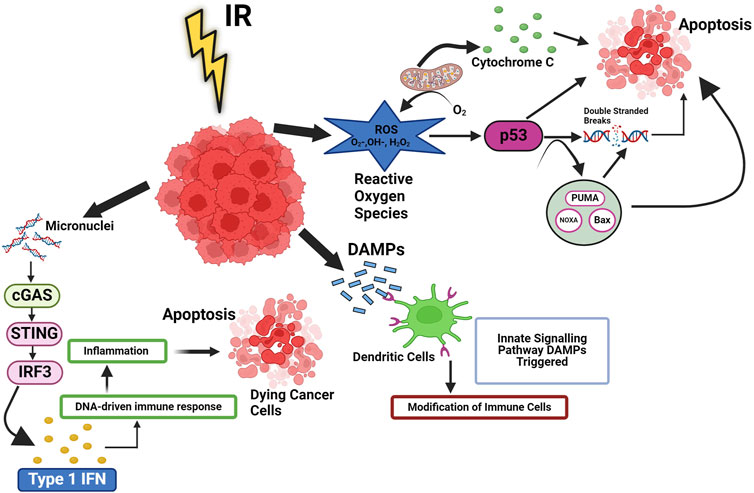
Figure 1. Ionizing radiation induced cellular pathways in cancer. Ionizing radiation (IR) trigger cell death, which leads to release of DAMPs and cytokines, then activate innate signalling pathways and modification of immune cells. These signals improve the processing of TAAs and the cross-presentation of antigenic peptides to CD8+ T lymphocytes via MHC I. They also favour the recruitment of APCs like DCs, the uptake of dying tumor cells, and the processing of TAAs. Additionally, radiation can cause the release of type-I IFN from immunological and cancer cells as well as activate the complement system, which can increase the activation of both DC and T cells. RT can also result in MHC I upregulation. p53 signalling is linked to the reactive oxygen species (ROS) response to radiation. Radiation-induced mitochondrial damage helps irradiation boost intracellular ROS levels. When ROS levels are high, p53 may significantly reduce the oxidative damage imposed on by radiation. Type I IFN is produced when cytoplasmic chromatin DNA stimulates the cGAS-STING pathway and regulates DNA derived immune response.
IR directly causes DSBs while also causes base damage through indirect effects. Moreover, IR also leads to the formation of ROS, which indirectly contributes to DNA damage. These ROS cause DNA to develop apurinic/apyrimidinic (abasic) sites, SSBs, changes to the sugar moiety, and deaminated adducted bases (Srinivas et al., 2018). When DNA sustains damage, the cell’s repair machinery is activated, prompting the halting of the cell cycle at specific control checkpoints. This pause allows the cell to undertake the repair of the DNA damage, thereby preventing the cell cycle from progressing further.
ROS can induce various types of DNA damages, including generation of abasic sites, SSBs, chemical modifications to the sugar moiety, and deaminated adducted bases. When DNA damage occurs, the cell activates repair mechanisms and halts the cell cycle at specific checkpoints to facilitate DNA repair. This response is crucial for preventing the propagation of mutations and maintaining the genomic stability (Davalli et al., 2018).
In the context of RT, if tumor cells possess efficient DNA repair mechanisms, they can develop resistance to radiation by repairing the damage induced by IR. This resistance allows tumor cells to survive and continue replicating despite the radiation treatment. However, if the DNA damage remains unrepaired or is too severe, the cell may undergo programmed cell death, known as apoptosis, to prevent the transmission of mutations to daughter cells (Curtin, 2023).
High doses of radiation can lead to toxicity and diminish the patient’s prognosis. Therefore, tailoring radiation treatment based on the individual’s DSB repair capability may help predict the toxicity to surrounding tissues, thus enhancing treatment safety (NCBI, 2024a). The DNA repair capacity holds important significance in determining the suitable treatment strategy for cancer patients, and functional tests can offer valuable insights for making these clinical decisions.
It is well known that IR directly induces DNA damage in cancer cells, which leads to the activation of systemic IR-induced signalling cascade leading to changes in tumor microenvironment, These changes tend to influence the tumor microenvironment and make the tumors much more receptive to IT by aiding the release of tumor antigens, which can be targeted by IT (Sharabi et al., 2015) by increasing the density of TILs (Tumor-infiltrating lymphocytes). It has also been shown that IR leads to cyclic GMP-AMP synthase (cGAS) activation, a stimulator of the interferon gene (STING) pathway, leading to the production of proinflammatory signals (McLaughlin et al., 2020) that have a bearing on the fate of cancer cells and tumors. However, it is important to note that there have been few reports that suggest that IR gives rise to immune suppression as well by modulating myeloid-derived suppressor cells (MDSCs) and regulatory T cells (Tregs) (Rodríguez-Ruiz et al., 2018). In the irradiated tumor microenvironment, several intracellular signalling pathways are also modulated by IR in cancer cells. Radiation induces the death of cancer cells primarily by inducing DSBs and the generation of ROS, as shown in Figure 1, which modulate the intracellular tumor microenvironment (Dagar et al., 2023). The formation of micronuclei is a phenomenon that occurs upon exposure to IR, which further leads to activation of the cGAS-STING pathway, leading to the production of type 1 interferon (IFN). In addition to nuclear DNA, mitochondrial DNA damage is also recognized by cGAS, which then forms a complex with DNA (NCBI, 2023). This leads to the formation of the second messenger cGAMP, which modulates downstream signalling for the production of type 1 Interferons, leading to the maturation of dendritic cells (DCs), as observed by an increase in 1) the expression of co-stimulatory molecules and 2) the migratory capacity of DCs (Sprooten et al., 2019). The fragmented cancer cells are exposed to IR release factors like damage-associated molecular patterns (DAMPs), which influence the functioning of different immune cells (Figure 1). NF-κB pathways (canonical and non-canonical) also play an important role in IR-induced responses. Beside cGAS-STING pathways, the NF-κB pathway is commonly mediated through the IKK dependent canonical pathway. Which works with IRF-3 to optimise the expression of the IFN-β gene (Garland et al., 2022). The association of IRF3 and NF-κB is essential for activating type I IFN in DCs, which are stimulated by irradiated tumor.
Although RT-induced DSBs are the most effective molecular events for damaging and killing cancer cells, the DNA damage repair capabilities inherent in cancer cells can lead to resistance and diminish the efficacy of therapy. This radiation induces various types of DNA damages, including complex DSBs, which are pivotal in eliminating tumor cells. RT employs fractionated doses of IR to exploit this property, generating significant levels of clustered DNA damage that challenge tumor cells’ repair mechanisms and impede their survival chances.
Additionally, the effectiveness of RT is augmented by adjuncts that enhances the sensitivity of hypoxic cells, which are often resistant to radiation treatment. These adjuncts capitalise on vulnerabilities within tumor cells’ DNA repair pathways, such as deficiencies in repair pathways like HR due to mutations in BRCA1 resulting in repair deficiency. Tumor cells with compromised repair mechanisms are more susceptible to the DNA damage induced by RT, leading to their targeted destruction (Deckbar et al., 2011). However, it is crucial to consider the potential impact of lower radiation doses on normal tissues. While tumor cells may receive cytotoxic doses, normal cells may experience non-DSB clustered DNA damage even at lower radiation doses. For instance, doses as low as 10–100 cGy have been observed to induce clusters of apurinic/apyrimidinic (AP) sites in primary human fibroblasts (Guo et al., 2011). This emphasises the necessity of striking a balance between the therapeutic benefits of RT for the cancer cells and the potential risks to the surrounding normal tissues. The treatment planning in RT aims to maximise the radiation dose to tumor cells while minimizing possible exposure to the surrounding healthy tissues, thereby reducing the likelihood of adverse effects. Ongoing research endeavours to refine RT techniques and develop adjunct therapies to enhance tumor cell eradication while safeguarding normal tissues from radiation-induced harm (Guo et al., 2011). Therefore, focusing on inhibiting DNA damage repair mechanisms, such as by inhibiting NHEJ or by targeting single-strand break repair (SSBR) and BER pathway, presents a promising therapeutic approach to enhance the sensitivity of cancer cells to RT, offering a precise and effective treatment strategy for cancer patients. The SSBR and BER pathways are responsible for repairing damaged bases and SSBs in DNA. Inhibiting BER/SSBR could result in unrepaired damage, which may convert to DSBs when encountering a replication fork. Thus, in cells that are already HR-deficient, like breast or ovarian cancer tumors (BRCA−/−), PARP inhibitor-induced BER suppression results in unrepaired double-strand breaks and eventual cell death (Abbotts and Wilson, 2017).
Another molecule that can effectively target the DNA repair pathway is DNA-PK, a critical enzyme in the NHEJ pathway, belongs to the PI3K family and plays a critical role in various cellular processes. Selective inhibitors of DNA-PK have demonstrated radio sensitization in preclinical investigations (Shinohara et al., 2005; Davidson et al., 2013; Ismail et al., 2004) Currently, three phase 1 clinical-trial are underway to evaluate the safety and tolerability of a DNA-PK inhibitor (M3814) in combination with palliative RT with or without IT for advanced solid tumors (NCT02516813 and NCT03724890), as well as in combination with curative-intent RT for locally advanced rectal cancer (NCT03770689).
IR-induced DNA damage remains a pivotal component of cancer therapy. Harnessing the molecular vulnerabilities arising from this damage, along with incorporating innovative treatment strategies, is critically important for advancing the field and achieving better outcomes for cancer patients. Advancements in understanding radiation-induced DNA damage and ongoing research into personalized treatment approaches hold promise for improving cancer therapy outcomes. Exploring novel targets and refining treatment modalities will likely contribute to more effective and less toxic cancer treatments in the future.
Combining DDR with chemotherapy (CT)Historically, the majority of traditional CT treatments, such as direct agents that damage DNA, have been regarded as immunosuppressive, and one of the most frequent adverse effects of cytotoxic CT that limits dosage is lymphopenia. On the other hand, a growing corpus of experimental and empirical data indicates that certain CT, when administered at recommended dosages, may stimulate immunogenic tumor cell death and influence the tumor microenvironment to support immunity against tumors (Galluzzi et al., 2017). Immunogenic cell death pathways are the first mechanism via which CT has been demonstrated to activate the host immune system. Contrary to apoptosis, which is usually thought to be non-immunogenic, CT may cause cell death and the release of antigens from tumor cells. CT-induced cellular stress increases the immunogenicity of the cell by encouraging the surface expression and secretion of damage-associated molecular patterns (DAMPs) (Klarquist et al., 2014). Apart from cytosolic DNA, which is a potent DAMP, several other cellular proteins have also been observed to function as DAMPs. These include heat-shock proteins, calreticulin, hyaluronan, and high mobility group box 1 (HMGB1). DAMPs that have been released attach to receptors on stromal and cancer cells, triggering a host immune response that is like that of a pathogen. Type I IFN and other chemokines are secreted more readily when DAMP is activated, and DCs need these chemokines to activate tumor-specific CD8+ T lymphocytes (Diamond et al., 2011). Additionally, MHC class I and cancer-testis antigens can be expressed more prominently during DNA-damaging CT, while inhibitory mediators such as PD-L1 can be expressed less prominently on the surface of cancer cells (Vereecque et al., 2004). DNA-damaging CTs exert a profound impact on the tumor microenvironment, influencing immune regulatory cell activity and the tumor vasculature, thereby facilitating antitumor immunity. The term “antitumor activity” describes a treatment’s or intervention’s multidimensional capacity to obstruct a tumor cell’s ability to grow, spread, or survive via a variety of methods. This involves the direct destruction of tumor cells through the use of medications used in CT, targeted therapies, and IT, which cause cancer cells to undergo certain cell death processes such as necrosis or apoptosis (Khalaf et al., 2021). Additionally, interventions may hinder tumor cell proliferation and division by disrupting signalling pathways or inhibiting DNA replication, using repair enzymes. Tumor vasculature disruption, aimed at starving tumors of vital nutrients and oxygen by targeting their blood supply, is another strategy (Jing et al., 2019). Furthermore, antitumor immune responses can be activated through ITs, leveraging the body’s defences to identify and eliminate cancer cells or block immune checkpoints hindering antitumor responses. Finally, antitumor strategies may prevent metastasis by impeding the invasion and migration of cancer cells to distant sites. The overarching aim of antitumor approaches is to effectively combat cancer while minimizing harm to healthy tissues, thereby enhancing patient outcomes and quality of life (Said and Ibrahim, 2023). CTs have been demonstrated to downregulate these inhibitory signals in a few contexts, and there are numerous feedback mechanisms that work to limit the host immune response. For example, it has been demonstrated in animal models that medications including gemcitabine, cyclophosphamide, paclitaxel, fludarabine, and 5-fluorouracil decrease Treg or myeloid-derived suppressor cell (MDSC) function (Lutsiak et al., 2005; Zhang et al., 2008). Upregulating DC function is another way to activate the immune system, and CT drugs like cyclophosphamide have been demonstrated to boost DC activity and quantity (Nakahara et al., 2010). These findings underscore the multifaceted effects of DNA-damaging CTs in modulating the tumor microenvironment to promote antitumor immune responses. Perhaps not surprisingly, given the interaction between DNA damaging drugs and the tumor immune response, several lines of research has shown thar cytotoxic CT sensitizes tumors to ICB. For instance, in a mouse model of lung cancer, pre-treatment of the animals with oxaliplatin and cyclophosphamide was sufficient to promote sensitivity to host T cell immunity (Wang et al., 2015). In ovarian cancer, decitabine improved lymphocyte activity and worked in concert with CTLA-4 inhibition.
Advancement in synergy between DDR and ITCancer IT represents a promising new therapeutic paradigm to harness the patients’ immune system to eliminate the cancer cells (Kalisz et al., 2019). Both immune evasion and genome instability are characteristic features of cancer. When a proto-oncogene or tumor suppressor gene experiences DNA damage and there are insufficient or no DNA repair mechanisms available to fix the damage, tumorigenesis is often triggered. Immuno evasion subsequently stops the host immune system from identifying these transformed cells. Cancer has proven to respond well to treatments that target immune evasion and genetic instability (Lee et al., 2022). Variations in DNA damage response genes and the resultant genomic instability have a significant role in determining the antigenicity of tumors via both neoantigen-dependent and independent processes (Li and Chen, 2018). As a result, there has been a growing focus on utilizing DDR mutational status as a predictive biomarker for the response to immune checkpoint blockade. This approach aims to enhance patient selection and guide therapeutic choices (Aiello et al., 2020). Similar to RT, DDR deficiency results in heightened DNA damage and an increased tumor mutational burden (TMB) due to the accumulation of point mutations and indels, a hallmark of cancer (Said and Ibrahim, 2023). The higher number of DDR mutations influences the efficacy of IT. Immune checkpoint blockade is a form of IT that prevents the activation of inhibitory immune checkpoints, enabling the immune response to target cancer cells (Dunn et al., 2004). The link between the host immune system and the tumor has been understood for several years, and the ITs aimed at inducing the host immune system to eliminate the tumor cells have demonstrated some degree of clinical effectiveness. For example, therapies include the use of systemic IL2 in metastatic melanoma, renal cell carcinoma, as well as intravesicular Bacillus Calmette-Guerin (BCG) in bladder cancer (Klapper et al., 2008). Nonetheless, the use of antibodies against inhibitory signalling molecules on tumour and immune cells in clinical trials has revolutionised the area of cancer IR in the last 5 years. Genomic instability carried on by DDR failures, can activate signalling pathways such as cyclic GMP-AMP synthase-stimulator of interferon genes (cGAS-STING), upregulate the expression of programmed death ligand 1 (PD-L1), and enhance the production of DNA-based neoantigens. In a small number of patients, ICIs such as anti-cytotoxic T-lymphocyte-associated protein 4 (anti-CTLA4) and anti-PD1/PD-L1 antibodies have significantly improved treatment outcomes. Ipilimumab (anti-CTLA-4), an immune checkpoint inhibitor, was the first treatment to show a benefit in survival for patients with metastatic melanoma (Mellman et al., 2011) (shown in Figure 2). Subsequently, there has been proof that therapies targeting the PD-1 pathway, such as PD-1/PD-L1 inhibitors, have shown strong therapeutic efficacy. PD-1 is an immune checkpoint expressed on T cells, while PD-L1 is found on tumor cells (Vereecque et al., 2004; Lutsiak et al., 2005; Zhang et al., 2008). By blocking PD-1/PD-L1 interaction, these therapies unleash the immune system to recognize and attack cancer cells, leading to significant improvements in patient outcomes across various cancer types (Borst et al., 2021). PD-L1 is a binding partner is expressed on antigen presenting cells such as tumour cells and DCs. T cell activation and proliferation are reduced when PD-1 binds to PD-L1 (Chen and Han, 2015; Freeman et al., 2000). PD-1/PD-L1 inhibitors have been extensively evaluated in clinical settings on a variety of cancer types, including colon, lung, and melanoma. The FDA has approved PD-1/PD-L1 inhibitors such as pembrolizumab, nivolumab, durvalumab, and atezolizumab for the treatment of cancer. To date, research has demonstrated that the PD-L1 expression in tumors is controlled by DNA repair and signalling via numerous routes. Endogenous DNA damage in tumors may be continuously produced by oxidative stress or aberrant cell cycling prior to cancer treatment (Katerji and Duerksen-Hughes, 2021). One such pathway involves the activation of interferon regulatory factors (IRFs) and nuclear factor kappa B (NF-κB) in response to DNA damage. These transcription factors can induce the expression of pro-inflammatory cytokines and chemokines, including interferons and interleukins, which promote an inflammatory microenvironment within the tumor. This inflammatory milieu can, in turn, upregulate the expression of PD-L1 on tumor cells, facilitating immune evasion. Moreover, DNA damage-induced signalling pathways, such as the Ataxia-telangiectasia mutated (ATM) and ataxia telangiectasia and Rad3-related (ATR) pathways, can directly regulate PD-L1 expression through various mechanisms. For instance, activation of ATM and ATR kinases can lead to the stabilization of PD-L1 mRNA or the phosphorylation of transcription factors involved in PD-L1 transcriptional regulation (Kakoti et al., 2020). In such situations, immunological signalling may be upregulated by DNA damage responses. However, the immune activity under the circumstances without extra exogenous DNA damage, e.g., prior to RT/CT, is not totally able to overcome malignancies (Zhang et al., 2020). This condition will be expected in case of low TMB/MSI tumors. In contrast, recent studies have demonstrated that several immunological responses, including the release of interferons (IFNs, immune positive response) and PD-L1 upregulation (immune negative response) are generated after DNA damage-associated cancer treatments, such as RT and CT. DNA fragments accumulate in the cytoplasm of cells as a result of DNA damage, and cyclic GMP-AMP synthase (cGAS) is able to identify these fragments. cGAS binds to dsDNA sequences and begins signalling downstream through the STING pathway (Ye et al., 2021), which is a protein that affects immune system modulation. Type I interferon (IFN-1) and other inflammatory cytokines are expressed more when STING stimulates gene transcription via interferon regulatory factor 3 (IRF3) (Figure 3). Furthermore, STING can trigger a transcriptional response via the nuclear factor kappa-light-chain enhancer of activated B cells, including canonical and noncanonical pathways (Motwani et al., 2019). This leads to an increase in DCs presentation of tumor antigen, which in turn intensifies the CD8+ T-cell responses.
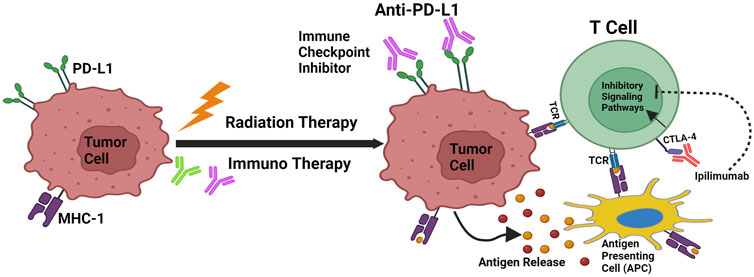
Figure 2. Cancer immunoediting and RT/IT Synergism. Mechanisms that support radiation and immunotherapy’s synergistic effects. Through antigen release, calreticulin activation, and CD47 downregulation, radiation increases the capacity of antigen-presenting cells to transmit tumor antigens to naive T cells. T-Cell Receptors (TCR) engage when MHC-1 is expressed, which leads to the antigen being presented. As PD-L1 and CTLA-4 are upregulated by radiation, immunotherapy that specifically targets these pathways can increase the effectiveness of radiation therapy.
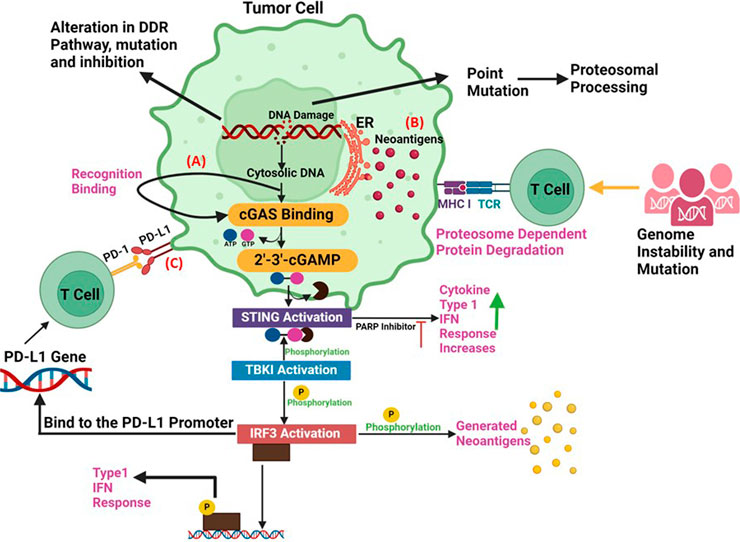
Figure 3. Synergy between DDR and Immunotherapy (A) The accumulation of cytoplasmic DNA and activation of the cGAS/STING pathway: DNA damage response defects can increase the cytosolic DNA., that can generate the xGAMP by activation of cGAS. By catalysing the synthesis of cGAMP, which functions as a second messenger in the activation of STING pathway, the cytosolic DNA sensor cGAS can trigger innate immune responses. When the STING pathway is activated, STING undergoes a conformational shift that causes an endoplasmic reticulum to perinuclear endosome shuttle. TBK1 could phosphorylate both IRF3 and STING, which increases the synthesis of type I IFNs. (B) Neoantigen Production: Alterations, mutations and inhibition in DDR pathway can promote the production of tumor neoantigens. Deficits in DDR produce neoantigens that enhance tumour identification. According to the neoantigen hypothesis, a non-synonymous mutation modifies an amino acid, resulting in the production of a new peptide. Therefore, the immune system can identify cancer cells lacking DDR as alien. (C) PD-L1 upregulation via DNA damage signals: The upregulation of PD-L1 expression is regulated by DNA damage signalling and DDR deficits. Immune checkpoint inhibitors are susceptible to cancer cells treated with PARP inhibitors. ICIs combined with PARP inhibitors show promising results against cancer since PARP inhibitors upregulate the expression of PD-L1 on cancer cells, promote genomic instability, and activate immunological pathways.
There are several molecular mechanisms that result in the synergy between IT and RT, improving their combined therapeutic efficacy. By causing immunogenic cell death, RT releases tumor associated antigens, which in turn stimulate T cells and DCs responses. Additionally, it alters the tumour microenvironment by boosting immune cell infiltration and lowering immunosuppressive factors (Yu S. et al., 2022). Type I interferon production is increased upon activation of the cGAS-STING pathway by RT-induced cytosolic DNA, hence augmenting anti-tumor immunity. Additionally, by enhancing DNA damage and boosting the generation of ROS, as well as acting as immunomodulator delivery vehicles, nanoparticles like gold and hafnium oxide augment the effects of RT (Yu R. et al., 2022). Since ICIs like anti-CTLA-4 and anti-PD-1 prevent T cell depletion and maintain their anti-tumor effectiveness, the combination of RT and these agents has demonstrated great potential. These synergistic pathways demonstrate how RT and IT can work synergistically to enhance the immune system’s ability to fight cancer and improve treatment outcomes (Shiravand et al., 2022).
IT combined with DDR-targeted drugs can overcome immune suppression imposed on by DDR abnormalities in cancer cells, thereby establishing antitumor immune responses and enhancing therapeutic results. This synergistic approach has considerable potential to improve overall survival rates and long-term disease control in cancer patients by increasing the patient group eligible for treatment and preventing or delaying the formation of resistance to IT (Wang et al., 2022).
RadScopal effectResearchers are exploring a relatively newer concept of ‘RadScopal effect’ that is described as the immunomodulatory effect that uses both high-dose radiations for immune priming in the primary tumor alongside low dose radiation targeted at the secondary tumor to support immune cell infiltration and promote effective tumor eradication. James Welsh proposed this radiation approach by combining high dose RT (HDRT) with low dose RT (LDRT) (Barsoumian et al., 2020a). Low dose radiation may be an effective tool to overcome the limitations of immunosuppressive factors produced by conventional RT. The fundamental mechanisms underlying the discernible impact of low-dose RT might be initiated by the initiation of DNA damage (Mavragani et al., 2015). Studies have shown that low dose RT creates a welcoming environment for the immune cells and promotes an anti-tumor response. Klug and co-workers showed that LDRT shifts pro-tumor M2-macrophages towards the anti-tumor M1-phenotype, increases the infiltration of CD4+ T cells and Natural Killer cells, and downregulates the expression of the inhibitory cytokine TGF-β (Klug et al., 2013). A proteomic analysis shows the role of LDRT in the upregulation of stimulatory factors and tumor microenvironment specific cytokines. MIP1α, CD137 (4-1BB), and Granzyme B were upregulated in tumor-infiltrating CD4+ T cells, indicating their activation and effector function. Also, in murine lung cancer models, LDRT further enhanced the effectiveness of CPIs such as anti-PD1 and CTLA-4, as confirmed by diminished tumor growth rates and prolonged survival (Patel et al., 2021). A recent clinical study was done in which it was found that, in patients with immune-resistant solid tumors, low-dose RT + high-dose RT safely enhanced lesion-specific responses by enhancing the infiltration of immune cells into the tumor microenvironment. Another study done by Herrera et al. endorsed the reasoning behind integrating LDRT with IT for metastatic ovarian cancer (Klug et al., 2013).
Although the radscopal effect, in which targeted radiation therapy incites a systemic immune response against tumors, has great potential for treating cancer, its practical implementation is restricted by various issues. This is a quite uncommon and erratic phenomenon that varies in its occurrence depending on the type of tumor and the environment. Tumors frequently develop immunosuppressive mechanisms that can reduce the efficiency of the radscopal effect, and the best radiation dosages and fractionation schedules to produce it are still unknown. The clinical usefulness of RT is further complicated by difficulties in tracking and evaluating its systemic effects (Wang et al., 2023).
Clinical benefits of combining RT with ITThe clinical benefits of combining ITs with RT has been demonstrated by many pre-clinical studies. The conventional benefits of adding CT with RT also hold ground for IT addition. RT and IT have been shown to have synergistic benefits by virtue of, the supra-additive effect of the two modalities. To achieve a certain biological effect, lower individual doses are required. Radio-sensitizing properties of IT agents will act on the same principles (Weichselbaum et al., 2017) as well as spatial cooperation. The combination of radio and IT can help overcome the treatment resistance. Tumor stroma and associated macrophages cause T-cell anergy and inefficient T-cells migration into the tumor. It is considered as one of the major reasons behind the resistance to IT (Turgeon et al., 2019). Targeted activation of the tumor microenvironment by combining with RT may overcome this resistance and augment the clinical outcome (Sharma et al., 2017; Waldman et al., 2020). A combination of RT and IT may prevent disease recurrence, as combining them may induce protective “immunologic memory,” which in turn could prevent disease recurrence.
As of now, our understanding of the intricate relationship between radiation and the immune system remains incomplete, yet numerous intriguing observations have emerged. The cytotoxic impact of RT on tumor cells facilitates the generation of tumor neoantigens for T lymphocytes and triggers the release of pro-inflammatory cytokines, thereby fostering an immune response. In preclinical and clinical settings, various groups used the advantageous immunomodulatory properties of radiation to initiate a more potent systemic anticancer immune response against tumors throughout the body (Morris et al., 2016). This treatment approach, in situ vaccination, where the patient’s tumor serves as a source of tumor-specific antigens, prompting and broadening a robust antitumor T cell response. Many clinical findings justify combining radiation with immune checkpoint blockade (Deng et al., 2014; Dovedi et al., 2017). Different studies highlight the use of IR/IT in different types of cancer including melanoma, NSCLC, rectal cancer, etc., as shown in Table 1 (Chen D. et al., 2020). When compared to monotherapy, IRT offers promising avenues for enhancing localized lesion control and inducing the abscopal effect. Here, we review the recent IRT clinical trials and delve into their potential significance in clinical use. Initially, Golden and his team demonstrated that the combination of local RT and granulocyte-macrophage colony-stimulating factor (GM-CSF) indeed yields a substantial objective abscopal effect in individuals afflicted with metastatic solid tumors (Golden et al., 2015).
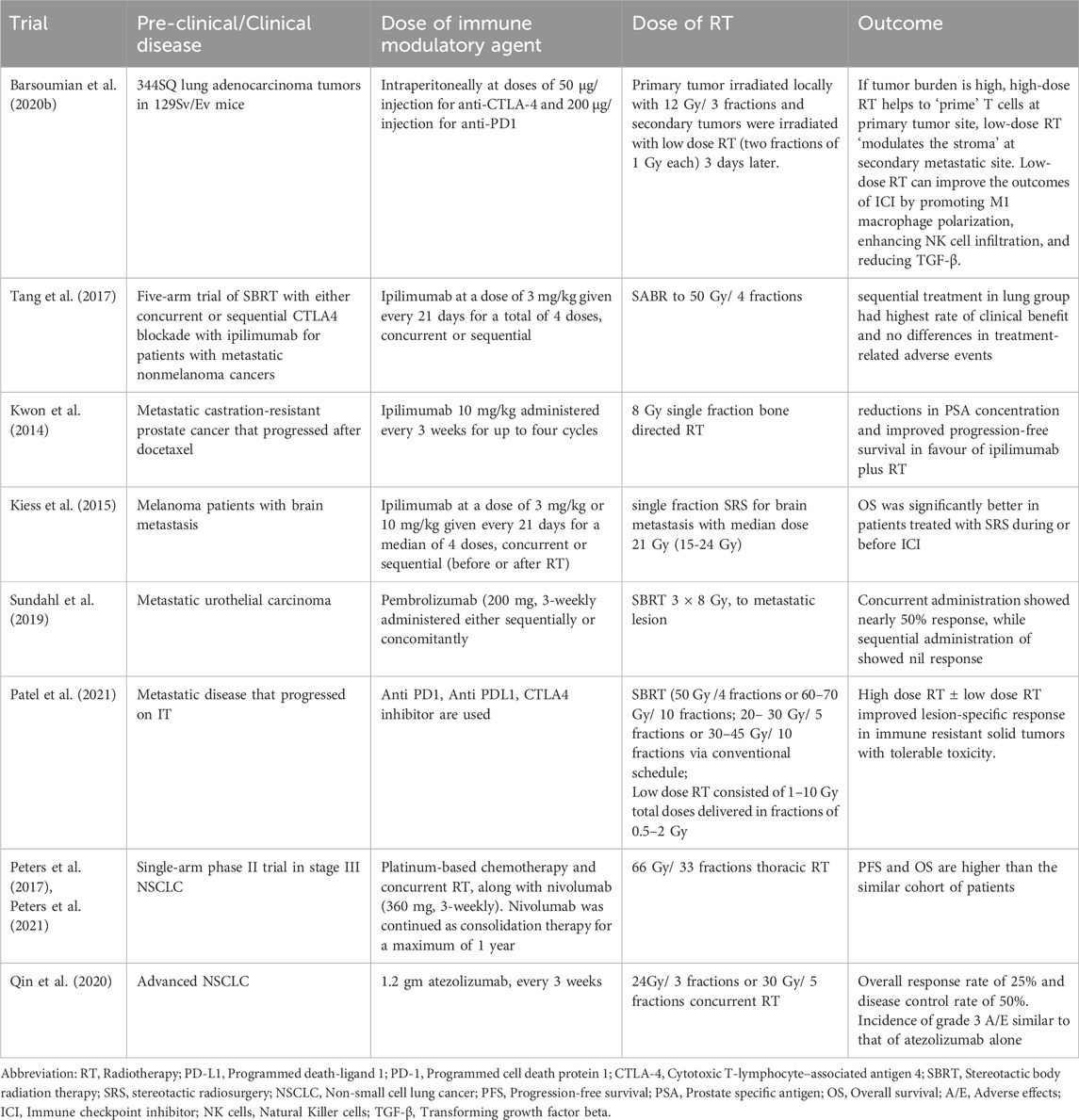
Table 1. Pre-clinical and clinical trials using various immunotherapy and different radiation schedule.
A Phase 1 study was conducted to evaluate the immunologic response and safety induced by autologous DCs in hepatoma patients who received a single fraction of RT. Out of 14 patients, two patients had achieved a partial response (Chi et al., 2005). Shaverdian and co-workers performed a secondary examination of the KEYNOTE-001 trial, revealing that individuals who underwent a combination of RT and pembrolizumab had longer progression-free survival and improved overall survival compared to those who had not undergone prior RT while maintaining a reasonable safety profile (Shaverdian et al., 2017). A prospective trial has been conducted to show that the combination of RT with immune checkpoint blockers has a significant role in the survival of patients (shown in Table 2). Kwon et al. conducted a multicentre phase 3 clinical trial with men who had at least one bone metastasis from castration-resistant prostate cancer that had advanced following docetaxel treatment, based on good preclinical findings in a spontaneous mouse model of prostate cancer. Patients were given either ipilimumab or a placebo after radiation therapy (8 Gy in one portion) for bone metastasis. Comparing ipilimumab to placebo did not affect overall survival (p = .053) (Kwon et al., 2014). Patients with good prognostic indicators (no visceral metastases, no anaemia, normal alkaline phosphatase) and treated with ipilimumab exhibited a statistically significant enhancement in survival relative to those receiving the placebo (Kwon et al., 2014). A study conducted by Susan M Domchek et al., focused on patients with germline BRCA1-or BRCA2-mutated metastatic breast cancer, an open-label, multicenter, phase 1/2 basket study was designed to evaluate the safety and efficacy of olaparib in combination with the PD-L1 inhibitor durvalumab. The multicentre, open-label, phase 1/2 MEDIOLA pilot project is evaluating durvalumab with Olaparib in solid tumours. The four groups that were enrolled were germline BRCA-mutated metastatic ovarian cancer, relapsed small-cell lung cancer, germline BRCA-mutated metastatic breast cancer, and metastatic gastric cancer. The cohort with breast cancer is the subject of this summary. Individuals with progressive, HER2-negative metastatic breast cancer who were at least 18 years old (or 19 years old in South Korea) and had germline BRCA1 or BRCA2 mutations were included. After receiving 300 mg of Olaparib twice day for 4 weeks, participants were given 300 mg of Olaparib plus 1.5 g of durvalumab every 4 weeks until the disease progressed. Safety, tolerability, and the 12-week disease control rate were the main outcomes. The study is still underway even though recruitment is over (ClinicalTrials.gov, NCT02734004) (Domchek et al., 2020). In another study, ICIs have shown good results in combination with PARP inhibitors in ovarian cancer. Immunogenomic profiling and single-cell imaging were used on tumor samples from patients enrolled in the phase I/II trial (NCT02657889) integrating pembrolizumab and niraparib. In this study, single-cell spatial analysis demonstrated a significant connection between PD-L1+ macrophages, PD-L1+ tumor cells, and fatigued CD8+ T-cells. Additionally, geographical analysis of the two extreme responders revealed distinct clustering of exhausted CD8+ T-cells with PD-L1+ macrophages in the first extreme responder, and exhausted CD8+ T-cells with cancer cells harbouring genomic PD-L1 and PD-L2 amplification in the second extreme responder (Färkkilä et al., 2020). Nonetheless, the optimal synergistic effects between immune ICIs and RT, encompassing factors like sequence, targeted irradiation sites, dosage, and fractionation, necessitate further exploration and refinement.
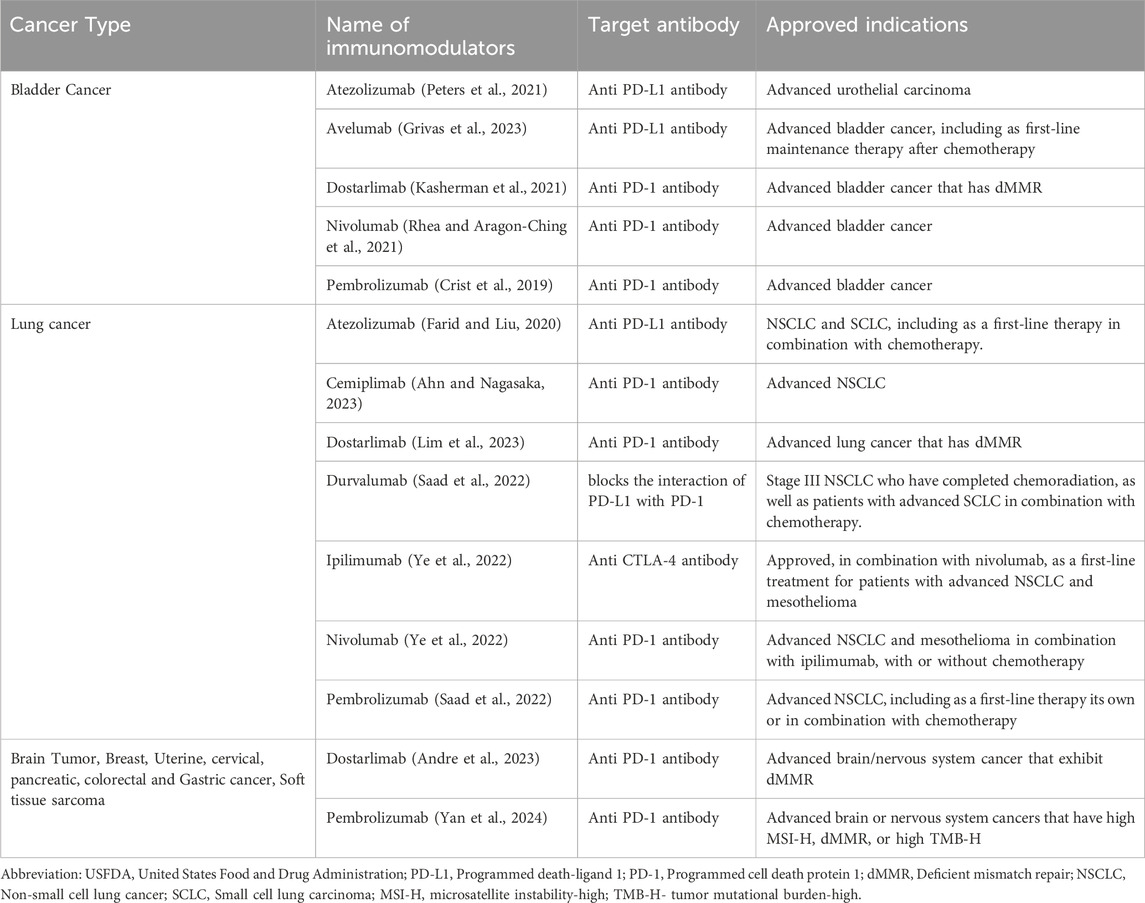
Table 2. Therapies and clinical indications of various USFDA approved immunotherapy agents.
Optimal RT dose-fractionation scheduleThe immune response induced by RT is “dose-dependent” and the optimum dose fractionation schedule is debatable. Several pre-clinical studies suggest higher doses per fraction can achieve better outcomes, maintaining low Treg numbers and a greater number of host immune cells infiltrating the tumors (Chen Y. et al., 2020). A series of clinical studies and reports also suggested that a combination of IT with stereotactic body radiation therapy (SBRT) produces excellent clinical results with tolerable toxicity (Chen Y. et al., 2020; Menon et al., 2019). In 2012, the first such case report demonstrated that ipilimumab in conjunction with SBRT (28.5 Gy/three fractions) produces a good local response at paraspinal metastatic mass but also regression of distant lesions away from the radiation field in a patient with metastatic melanoma (Tang et al., 2017)
A pooled analysis of two trials (PEMBRO-RT and MDACC) demonstrated higher doses of radiation along with pembrolizumab can significantly increase responses in patients with metastatic NSCLC, as shown in Table 2 (Reisz et al., 2014; Huang et al., 2013). In the PEMBRO-RT trial, pembrolizumab was given sequentially less than 1 week after the last dose of radiotherapy (24 Gy/three fractions), whereas in the MDACC trial, pembrolizumab was given concurrently with the radiotherapy (50 Gy/four fractions or 45 Gy/15 fractions). These results also corroborate the other similar clinical trials confirming the safety and efficacy of SBRT along with ITs. Nevertheless, few studies involving conventional doses of RT with IT showed no significant difference in outcome and hence can be effectively administered when SBRT is not feasible (Ferris et al., 2022; Elbers et al., 2020). The heterogeneity of tumor cells and their radiosensitivity probably contribute to differential responses to dose fractionation schedules of RT.
Selecting the optimal dose of RT involves considering factors such as tumor characteristics, treatment intent (curative vs palliative), radiation sensitivity, normal tissue tolerance, treatment schedule, patient factors, and clinical guidelines. Treatment planning tools and sophisticated imaging methods aid in ensuring that the tumor receives a precise dose while neighbouring healthy tissues are spared (Wang et al., 2023). Close monitoring during treatment enables adjustments based on toxicity and tumor response. Radiation oncologists optimize tumor control while minimizing side effects by incorporating these factors into the dose of radiation therapy that they administer to each patient.
Tumors may react differently to different RT schedules depending on differences in their radiation sensitivity and features. Radiation may more readily destroy some cells while sparing more resilient ones. Plans for radiation therapy can be adjusted to take these variations into consideration in order to maximize treatment efficacy and reduce harm to healthy tissue (Wang et al., 2023).
The optimal timing for RT in combination with ITThe selection of optimal timing of RT and the evaluation of the safety and efficacy of IT as “concurrent” or “sequential” warrant critical clinical judgment. Different types of IT target different immunological pathways, and radiation can exert its effect differentially based on the dose and fractionation. Therefore, a single strategy to achieve the greatest synergistic effects is not feasible for multiple cancers. IT drugs are typically administered concurrently or after RT to allow newly recruited T lymphocytes to effectively kill tumor cells both at the main site and at distant regions after being exposed to tumor antigens (Weichselbaum et al., 2017). The PACIFIC study demonstrated that Durvalumab® can be safely used as a maintenance medication with a survival benefit and limited toxicity following chemoradiotherapy (Spigel et al., 2022). However, a subset analysis of the PACIFIC trial suggests that initiating Durvalumab® within 2 weeks after completing chemoradiotherapy appeared to have greater progression free survival (PFS) rather than starting after 2 weeks of chemoradiation (Faivre-Finn et al., 2021). While sequential administration of IT followed by RT have been well established, many studies are enthusiastically investigating the effect of concurrent administration of IT. A phase II trial of concurrent Atezolizumab® with chemoradiation for patients with unresectable NSCLC, showed that concurrent RT with Atezolizumab® followed by the
留言 (0)