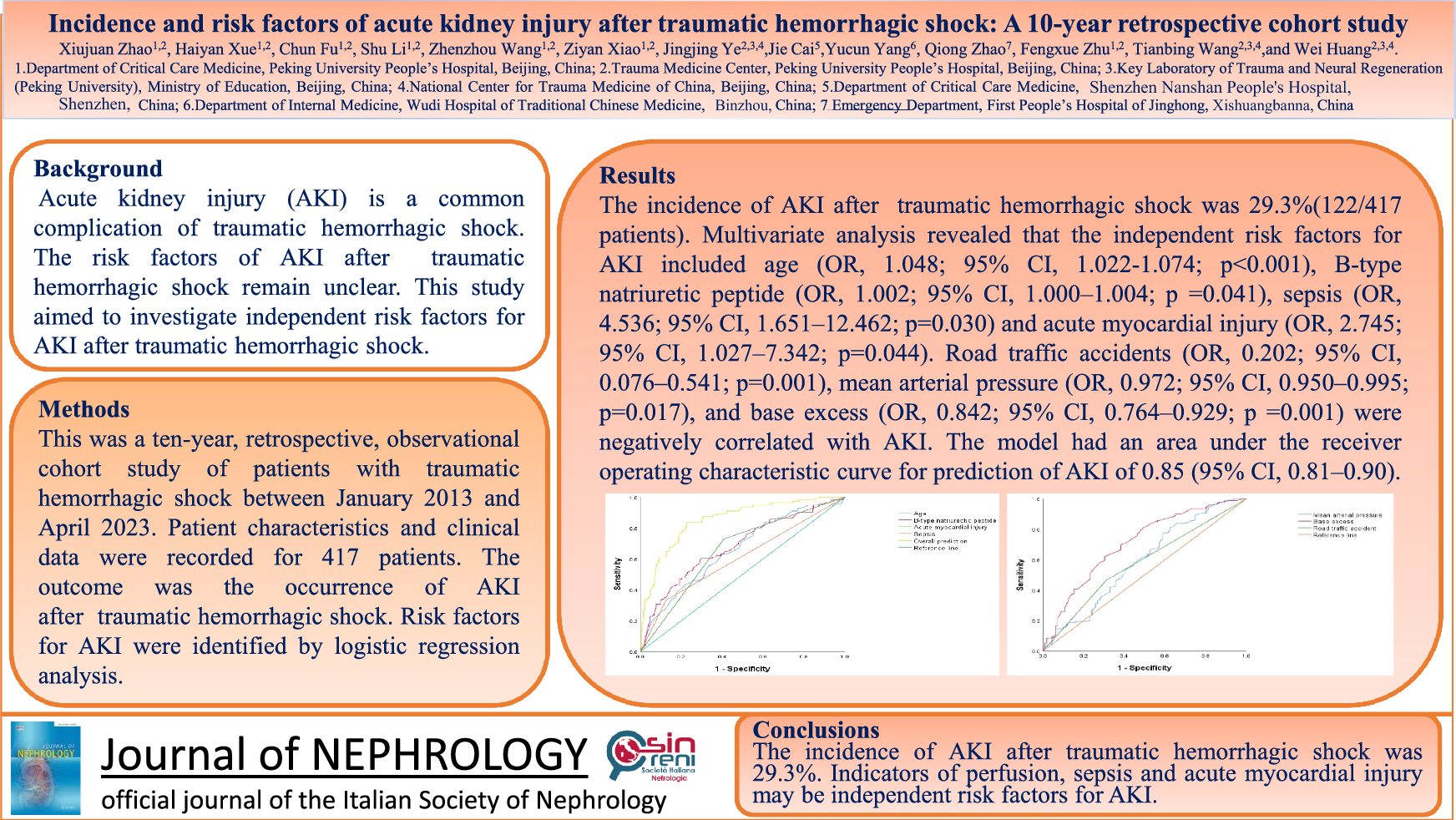
Herein we report a patient with a diagnosis of lysinuric protein intolerance who developed nephrotic syndrome accompanied by low C3 and C4. Immune complex-MPGN, lupus nephritis (LN), and C3 glomerulopathy were considered in the differential diagnosis before biopsy. The patient had no clinical findings compatible with systemic lupus erythematosus, except ANA positivity, and kidney biopsy was not suggestive of LN. The C3NeF was negative and there were deposits of co-dominant immunoglobulins/immune complex as well as C3 accumulation which excluded C3 glomerulopathy. Based on the laboratory and kidney biopsy findings, we diagnosed IC-MPGN and considered it to be related to lysinuric protein intolerance. Thereafter, she achieved remission with steroids and ACEi.
Lysinuric protein intolerance is a rare autosomal recessive metabolic disease caused by mutations in the SLC7A7 gene, disrupting the function of the cationic (y + L) amino acid transporter-1 (y + LAT-1), a transport protein of dibasic amino acids (lysine, arginine, and ornithine). Defective transport of these amino acids in the intestine and kidney leads to amino acid imbalances, disturbing multiple pathways, including protein synthesis and the urea cycle. A transport defect in non-polarized cells (lymphocytes or macrophages) is also likely implicated in the pathogenesis [2].
Lysinuric protein intolerance is a disease with multisystem involvement. Growth delay, recurrent fractures, hepatosplenomegaly, cirrhosis, diarrhea, hemophagocytic lymphohistiocytosis, pulmonary alveolar proteinosis (PAP), vascular lesions (cerebral infarction), pancreatitis are other clinical findings [2]. Kidney involvement may also be observed. Generally, it begins with mild proteinuria and hematuria and can progress to kidney failure. Tanner et al. described 39 patients with lysinuric protein intolerance with a mean age of 30.5 years (range, 1–62 years) and observed proteinuria in 74% and hematuria in 38%. Kidney failure developed in 4 patients, 2 of whom were < 25 years old [3]. Although kidney involvement is often tubulo-interstitial, glomerular involvement has been reported [4, 5].
The glomerular involvement in lysinuric protein intolerance ranges from lupus-like lesions to amyloidosis, according to published data [6]. Recent data from a series of five patients who underwent kidney biopsy between 1986 and 2015 mainly describe tubulointerstitial nephritis and nephrocalcinosis but one of the five patients showed a MPGN pattern [4]. Although immune complex glomerulonephritis (IC-GN) in lysinuric protein intolerance has been reported, in most cases this diagnosis was made on the basis of autopsy or kidney biopsy findings performed shortly before death. In a few cases, immunosuppressive treatment was attempted, but kidney response could not be assessed because the patients died shortly afterwards of various causes, including pulmonary alveolar proteinosis, hemophagocytic lymphohistiocytosis, septic shock, and multiorgan failure. To the best of our knowledge, there is only one patient whose response to immunosuppressive treatment for IC-GN has been documented in the literature. As in our patient, this patient presented with nephrotic syndrome and was followed for approximately 4 years. Initially, remission was achieved with steroid and mycophenolate mofetil (MMF) treatment, but later a relapse occurred after the steroid treatment was stopped, and therefore, steroid treatment was resumed. Since then, partial remission was achieved with persistent proteinuria (approximately 1 g/d) on steroid and MMF treatment [4, 5]. Our patient developed nephrotic syndrome and steroid treatment was initiated after the kidney biopsy. By the third month, the patient was in complete remission. Steroid treatment was tapered and discontinued after 18 months and she was followed for 3 years after the kidney biopsy and has been in remission without treatment for the last 17 months. This result supports that IC-MPGN, which is very rarely seen in patients with lysinuric protein intolerance, can benefit from immunosuppressive treatment.
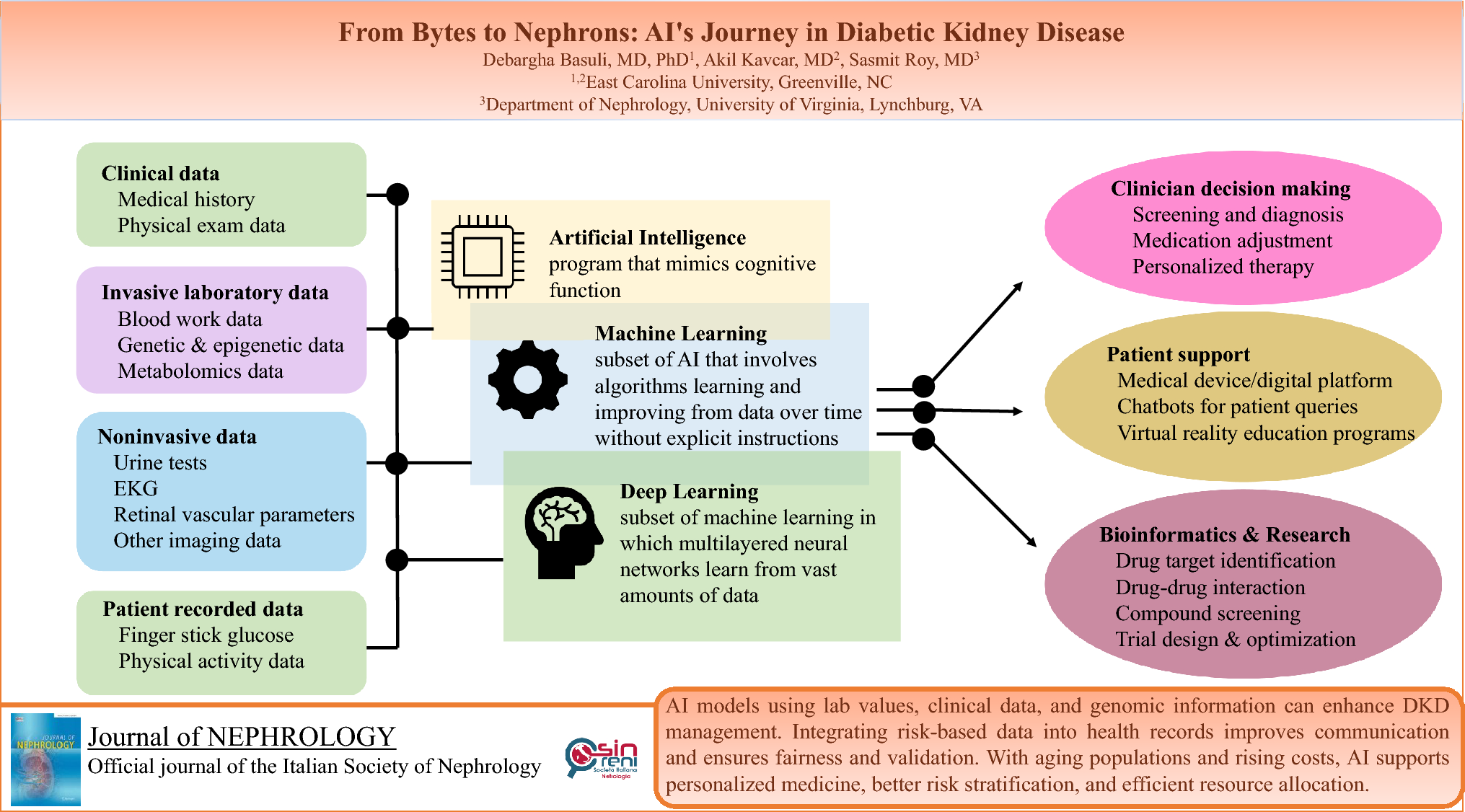
Immune dysregulation is a well-known complication in lysinuric protein intolerance that has been associated with hemophagocytic lymphohistiocytosis, autoimmune diseases, and pulmonary alveolar proteinosis. The underlying mechanism is still obscure. Involvement of the mononuclear phagocytic system appears to be the main factor. One of the culprits thought to be involved in the pathophysiology is the nitric oxide (NO) pathway. Nitrate oxide has significant effects on the immune system, cytokine production, and cyclooxygenase activation. Because of defective efflux through the cationic amino acid transporter, arginine accumulates in the monocytes/macrophages of lysinuric protein intolerance patients, causing increased NO synthesis. Manucci et al. [7] reported increased plasma NO levels in patients with lysinuric protein intolerance. Therefore, it was hypothesized that increased NO is a major initial trigger of activation of the immune system and immune abnormalities in lysinuric protein intolerance patients. On the other hand, Rotoli et al. [8] showed increased inflammation in SLC7A7-silenced monocytes, that was independent of intracellular arginine concentration.
In conclusion, although tubular dysfunction, nephrocalcinosis, and chronic tubulointerstitial nephritis are the most common forms of kidney involvement in lysinuric protein intolerance, glomerular involvement with immune-mediated glomerulonephritis may also occur, and immunosuppressive therapy may be indicated.









留言 (0)