
記住我








The pancreas arises from a small cluster of cells in the embryonic gut tube during development (Joglekar et al., 2007). This process is tightly regulated by a complex network of signaling pathways and transcription factors, which control pancreatic progenitor cell specification, proliferation, and differentiation (Jarc et al., 2023). This glandular organ comprises different cell types, including endocrine cells that produce hormones and exocrine cells that produce digestive enzymes (Overton and Mastracci, 2022). The proper development and function of the pancreatic cells are critical for maintaining health and preventing diseases, such as diabetes and pancreatitis (Karpińska and Czauderna, 2022). These conditions are associated with defects in the molecular pathways that regulate pancreas development and function (Thrower et al., 2008; Polireddy and Chen, 2016). However, there is limited knowledge on the precise mechanisms underlying these defects, and further research is still needed to better understand the complex molecular interactions within the pancreas.
Acute pancreatitis has a rising global incidence and is associated with significant morbidity and mortality. In severe cases, mortality rates can reach 30%–40% (Iannuzzi et al., 2022). The pathogenesis of acute pancreatitis is multifaceted and involves both local and systemic inflammatory responses. Epigenetic regulatory mechanisms play an important role in controlling the inflammatory cascade (Zhou et al., 2022). Various studies have investigated the role of epigenetic modifications in regulating inflammation and acute pancreatitis (Sandoval et al., 2016; Sun et al., 2021). DNA methylation is one of the most extensively studied epigenetic modifications in the context of acute pancreatitis. Studies have shown altered DNA methylation levels on genes involved in inflammation and oxidative stress in the pancreas during acute pancreatitis episodes (Natale et al., 2019; Sun et al., 2021). For instance, promoter hypermethylation of anti-inflammatory genes such as IL10 and SOCS3 has been associated with decreased expression and increased inflammation in the pancreas (Yin et al., 2015). Similarly, hypomethylation of pro-inflammatory NFKB1 promoter regions increases expression and enhances pancreas inflammation. Histone modifications have also been implicated in the pathogenesis of acute pancreatitis. For example, histone H4 acetylation in the IL-1β promoter region correlates with increased expression of this pro-inflammatory cytokine in the pancreas (Pedersen et al., 2022). However, further research is needed to fully understand the complex interplay between epigenetic regulators and the inflammatory cascade in acute pancreatitis, which could lead to the development of new therapies for this debilitating disease.
Euchromatic histone-lysine N-methyltransferase 2 (EHMT2/G9a) is a methyltransferase that catalyzes the mono- and dimethylation of lysine nine on histone H3 (H3K9me1/2), leading to transcriptional repression. EHMT2 has been implicated in regulating various cellular processes beyond gene expression, including cellular differentiation and DNA repair (Jan et al., 2021). While EHMT2 directly regulates the differentiation and function of various immune cell types, including T cells, B cells, and macrophages, several studies also support the role of EHMT2 in non-immune cells to control inflammatory responses (Scheer and Zaph, 2017; Mourits et al., 2021). For instance, EHMT2 represses the expression of pro-inflammatory cytokines, such as TNF, in tumor cells to promote breast cancer recurrence (Mabe et al., 2020). Studies on vascular smooth muscle cells have also implicated EHMT2 in attenuating the IL-6 inflammatory response in atherosclerotic lesions (Harman et al., 2019). Furthermore, liver-specific Ehmt2 knockout leads to an enhanced proinflammatory response in lipopolysaccharide (LPS)-induced liver injury model (Lu et al., 2019). Recently, we have shown that Ehmt2 inactivation antagonizes oncogenic Kras-mediated pancreatic cancer initiation and promotion by altering growth and immune gene expression networks (Urrutia et al., 2021). Ehmt2 knockout driven by either Pdx1-Cre or P48Cre/+ demonstrated that this pathway is not required for pancreas exocrine development and is tolerated in this organ under basal contexts. However, the impact of Ehmt2 on the transcriptome during early development and under the inflammatory stressor of acute pancreatitis remains unknown.
Here, we investigate the role of Ehmt2 in postnatal murine pancreas development and caerulein-induced acute pancreatitis. Ehmt2 inactivation results in distinct transcriptional landscapes during pancreatic maturation, suggesting an increased susceptibility of this organ to an injury-inflammation-repair process. Congruently, in response to the induction of acute pancreatitis, we show that Ehmt2 inactivation leads to a more aggressive inflammatory response. Thus, this study advances our understanding of the epigenetic regulation and molecular mechanisms that play a role in pancreatic development, homeostasis, and diseases.
2 Materials and methods2.1 Mouse models and acute pancreatitis inductionAnimal care and all experimental protocols were reviewed and approved by the Institutional Animal Care and Use Committees of Mayo Clinic Rochester (IACUC protocols A00002240-16 and A24815) and the Medical College of Wisconsin (AUA00005963). Mice were maintained in standard housing with controlled temperature, humidity, and light cycles and given standard rodent chow and water ad libitum. Tissues were collected and preserved in formaldehyde for a minimum of 24 h prior to transfer to 70% (v/v) ethanol for histological processing and examination. B6.FVB-Tg(Pdx1-Cre)6Tuv/J (Pdx1-Cre, IMSR Cat# JAX:014647, RRID: IMSR_JAX:014647) (Hingorani et al., 2003) and Ptf1aTM1(cre)Hnak/RschJ (P48Cre/+, IMSR Cat# JAX:023329, RRID: IMSR_JAX:023329) (Nakhai et al., 2007) were originally purchased from Jackson Laboratories. Ehmt2 flox/flox (Ehmt2fl/fl) animals were generously provided by Dr. Oltz (Tachibana et al., 2007). Animals were maintained on a C57Bl/6 background, and genotyping procedures to confirm Pdx1-Cre;Ehmt2fl/fl and P48Cre/+;Ehmt2fl/fl crosses have been described previously (Urrutia et al., 2021). Both sexes were included in the experiments. Animals that were used in ontogeny studies with no additional treatments were sacrificed at 10 days (postnatal, PN) or 4 weeks (young adult, YA). For induction of acute pancreatitis, cohorts of 4-week-old mice were fasted for 12 h before the first caerulein injection in accordance with prior studies (Ding et al., 2003; Vasseur et al., 2004; Strobel et al., 2007; Shigekawa et al., 2012; Choi et al., 2015; He et al., 2024). Caerulein or saline control was administered via IP injection at a dose of 50 μg/kg once an hour for a total of eight injections. Food was returned after the first dose. Animals were euthanized 18 h after the initial dose. Tissue was taken (n = 2-4) for RNA and histological analysis. Mice were euthanized using CO2 in accordance with institutional guidelines.
2.2 Serum analysisBlood was collected from the animals by orbital puncture and serum was isolated for studying serum chemistry. Serum was mixed 1:1 with saline and their profile consisting of Glucose, Albumin, Globulin, Total Protein, Alanine Amino Transferase, Total bilirubin, Minerals (Sodium, calcium, Phosphorus) Blood Urea Nitrogen (BUN), and Alkaline phosphatase were measured using VetScan VS2 (Mathison et al., 2013).
2.3 Histological analysisPancreatic tissues were paraffin-embedded for sectioning, and sections were stained with hematoxylin and eosin (H&E) for pathological evaluation. Pancreatitis severity was assessed by assessing pancreatic tissue edema, inflammatory cell infiltration, and necrosis. The scoring system was adapted from (Moreno et al., 2006). It included an assessment of pancreatic tissue edema (on a scale from a minimum of 0 for no edema to a maximum of three for separated and disrupted acini), inflammatory cell infiltrate (on a scale from a minimum of 0 for no infiltrate to a maximum of 3 with infiltrate in the parenchyma for >50% of the lobules), and necrosis (on a scale from a minimum of 0 for absence of necrosis to a maximum of 3 with diffuse parenchymal necrosis for >10% of the parenchyma). To quantify the severity of the parameters, four random fields (×20 objective) per slide, each containing at least 1,000 cells per field, were imaged and assessed by the three scales. Mean ± SD scores were calculated for each parameter.
2.4 TUNEL assayFormalin-fixed pancreatic tissues were paraffin-embedded and sectioned (5 µm). TUNEL analysis was carried out using the ApopTag Peroxidase in situ cell apoptosis detection kit (Millipore, S7100) according to the manufacturer’s directions. Slides were developed with Nova Red (Vector Laboratories) and counterstained with Mayer hematoxylin. Five random fields (×20 objective) per section, containing at least 1,000 cells per field, were imaged and counted (Urrutia et al., 2020).
2.5 RNA extraction, RNA-seq, and bioinformatics analysisPreparation of RNA from tissue was performed as previously described (Urrutia et al., 2021). To reduce degradation during storage at −80°C, RNaseOUT (Invitrogen, Cat# 10777019) added to final elution. The RNA samples were quantified by Qubit (Invitrogen), and their quality was assessed using the Fragment Analyzer (Agilent). The samples with RINs >6 and DV200 > 80% were selected for library preparation. The pancreas RNA was then sequenced using the Illumina TruSeq RNA v2 library preparation kit and the Illumina High Seq-2000. The sequencing reads were mapped to the mouse reference transcriptome Gencode vM23 (GRCm38. p6), and at least 24 million mapped read pairs were obtained per sample. The resulting reads were processed through the Mellowes Center workflow, which includes MapRseq3 (Kalari et al., 2014) and EdgeR (McCarthy et al., 2012). Differential gene expression was based on a false discovery rate (FDR) < 0.1 and an absolute fold change (FC) ≥ | 2.0|. Pathway analysis of DEGs was done using RITAN (Zimmermann et al., 2019) and the MSigDB hallmark gene set collection (Liberzon et al., 2015), while gene network and upstream regulatory analyses were performed using Ingenuity® Pathway Analysis (IPA®; Qiagen). To quantify immune-cell fractions in the young-adult/postnatal bulk RNA-seq comparisons, we performed digital cytometry analysis with the quanTiSeq algorithms, which allowed intra-sample and inter-sample comparisons of only immune cell type fractions (Finotello et al., 2019). The quanTIseq method was applied on DEGs from through an R package called Immunedeconv allowing for identification of immune cell composition despite the low overall immune cell population (Sturm et al., 2019). Using the same R package for the bulk RNA-seq comparisons that involved caerulein injections, the MCP-Counter algorithm was used, which allowed for an expanded breakdown of the complete cell composition with robust quantification of the absolute abundance of eight immune and stromal cell populations (Becht et al., 2016).
2.6 Reverse transcription quantitative real-time PCR (RT-qPCR)Total RNA (2 μg) was used as a template for cDNA synthesis, using the RT2 First Strand Kit (Qiagen) according to manufacturer’s protocol. RT2 SYBR Green qPCR Mastermix (Qiagen) was used with the following reaction conditions: denaturation at 95°C for 10 min; 45 cycles of 15 s at 95 °C, 60 s at 60°C. Reactions were carried out using the CFX96 Real Time System, and real-time PCR data was analyzed using the CFX Maestro software v2.3 (Bio-Rad). Primer sequences for each transcript are provided in Supplementary Table S6.
2.7 Spatial transcriptomicsFFPE Pancreatic tissue sections (Ehmt2+/+ and Ehmt2fl/fl) were deparaffinized followed by H&E staining and imaging using BZ-X800 (Keyence). Thereafter, tissue was destained, decrosslinked, and permeabilized for processing with the CytAssist and Visium Spatial Gene Expression Kits (10x Genomics; Pleasanton, CA, United States). Library quality metrics were confirmed by fragment analysis and Kapa qPCR before sequencing according to manufacturer recommendations on the NovaSeq 6,000 (Illumina, San Diego, CA, United States). A sequencing depth of approximately 250–400 million read-pairs per sample was obtained. Sample processing, library preparation, and sequencing for this project was completed by the Mellowes Center for Genomic Sciences and Precision Medicine Center at the Medical College of Wisconsin (RRID:SCR_022926). The original read quality was checked by FastQC and FastQ Screen. Alignment, tissue detection, fiducial detection, and barcode/UMI counting were performed by Spaceranger Count. For read alignment, reference transcriptome was mouse mm10-2020-A with mouse Visium Probe_Set_v1.0. Seurat was used for downstream spatial data analysis. First, spots with zero counts were filtered out, and normalization was performed using the SCTransform method. Subsequently, dimension reduction and clustering were performed with PCA and UMAP analysis. Spatially expressed genes for each cluster were found using the default Seurat method. Spatially variable genes were found using two Moran’s I methods, specifically the Seurat clustering and UMAP. Results were integrated to Loupe Browser for data visualization. Each cell type was assigned to its respective cluster through manual review of the expression of a comprehensive set of marker genes, as per outlined by Zhou et al. (2022), and the analysis of genes of interest involved utilizing the scale value, either in the form of Log normalized or Feature sum.
2.8 Analysis of publicly available Ehmt2 ChIP-seq data generated across many cell typesTo confirm direct Ehmt2 target genes, we compared the results of RNA-Seq with several EHMT2 ChIP-Seq experiments, using the ChIP-Atlas interface (Zou et al., 2022). Briefly, RNA-seq values were correlated with the presence of peaks within a window of −/+10 kb from the transcription start site of the gene, using both the hg38 and mm10 reference genomes. The data was numerically harmonized with values transformed from continuous to digital because they originated from the different cell types, with the following identifiers (SRX number corresponds to the sample identifier): HEK293: SRX738354, SRX973412, SRX973413, SRX973414, SRX973415, SRX973428, SRX973429; A549: SRX3010248, SRX3010249; Hep G2: SRX10478057, SRX10478058; IMR-32: SRX16322836; K-562: SRX5457324, SRX5457325; LNCAP: SRX17413109, SRX17413110; MCF-7: SRX7030880, SRX7030881, SRX7030882, SRX7030883; RD/18: SRX5316603; RH-41: SRX4561210; Cardiomyocytes: SRX2497443, SRX2497444, SRX2499660, SRX2499661, SRX2499669, SRX2499670; EpiSC: DRX013321; and ES cells: SRX348394, SRX348395.
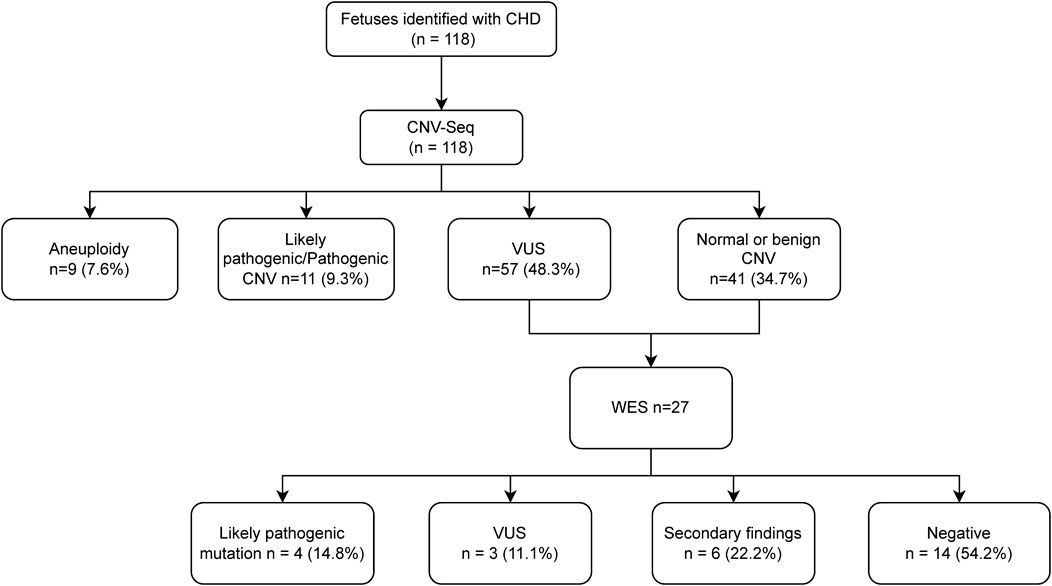
3 Results3.1 Loss of Ehmt2 increases the propensity of the normal pancreas to injury-inflammationPreviously, we reported that mice with pancreas-specific Ehmt2 knockout develop and grow normally, adopting the right size, shape, and histology (Urrutia et al., 2021). While this phenotype indicates that this protein is not essential for organ development, it does underscore a function for Ehmt2 in regulating gene expression networks during pancreatic maturation and pancreatitis. We harvested pancreas from Pdx1-Cre; Ehmt2fl/fl (Ehmt2fl/fl) and Pdx1-Cre;Ehmt2+/+ (Ehmt2+/+) mice at both postnatal (PN) day 10 and young adulthood (YA) 4–5 weeks of age and performed RNA-seq. Principal component analysis (PCA) shows a defined clustering of PN and YA Ehmt2fl/fl animals apart from their control groups (Figure 1A), which is more pronounced when the age groups are analyzed separately (Supplementary Figures S1A, B). We identified 143 and 125 unique differentially expressed genes (DEGs) between Ehmt2fl/fl and Ehmt2+/+ for YA and PN, respectively, with 26 overlapping between both age groups (Figure 1B; Supplementary Table S1). We performed network enrichments to infer the function of the transcriptional landscape of the pancreas from these animals (Figure 1C). Upregulated genes showed significant enrichment for fibrin clot formation, hypoxia, and KRAS signaling up in the PN Ehmt2fl/fl mice compared to those with Ehmt2 intact. The YA group was also enriched for these pathways and others, including erythrocyte gas exchange, extracellular matrix formation, and the scavenger receptor pathway. In contrast, no significant pathway enrichment was found among the downregulated DEGs. Comparison of molecular signatures in the YA upregulated DEGs group using MSigDB revealed enrichment of growth inhibitory pathways like P53 combined with KRAS signaling up, a phenomenon known to result in replication stress, cell cycle arrest, and apoptosis. More specifically, we found upregulation of key kinases involved in the P21 pathway, such as Cdkn1a, Chek2 and Ccng1, which we have previously shown causes cell-cycle arrest in the presence of activated KRAS in acinar cells (Urrutia et al., 2021). Moreover, these changes were accompanied by hypoxia, angiogenesis, and epithelial-mesenchymal transition (Figure 1C). To evaluate fractions of specific immune cell types with the limited number of DEGs, particularly as these mice were not immuno-stimulated and thus expected to have a relatively low number of resident immune cells, we conducted a constrained deconvolution analysis using quanTIseq (Finotello et al., 2019). Even though this method cannot determine differences in absolute abundance of immune populations, this analysis revealed alterations in the relative proportions of certain immune cell types induced by Ehmt2 inactivation at different developmental times. While the PN Ehmt2fl/fl pancreas had a 55.3% increase in dendritic cell populations along with 42.1% and 13.2% decreases in neutrophil and NK cells, respectively, we found that the mature pancreas at the YA stage became less variable between Ehmt2+/+ and Ehmt2fl/fl, with a 7.5% increase in marker genes for dendritic cells, a 7.3% decrease in neutrophil markers, and a minimal 0.2% decrease in NK cells (Figure 1D). Thus, we found that loss of Ehmt2 most notably results in de-repression or upregulation of genes, in particular those that relate to growth inhibitory programs and cellular stress responses.
Figure 1. Ehmt2 inactivation modulates the epigenetic landscape of mouse pancreas towards stress-related pathways. (A) Principal Component Analysis (PCA) based on Differentially Expressed Genes (DEGs) from RNA-seq conducted on pancreas tissue from mice at Postnatal (PN) day 10 and young adult (YA) 4–5 weeks, with and without Ehmt2, is shown. (B) Venn diagram illustrates the variation in DEGs upon Ehmt2 knockout (Ehmt2fl/fl) in the pancreas compared to Ehmt2 wild-type (Ehmt+/+) mice for YA and PN cohorts. (C) MSigDB Hallmarks and Reactome pathway enrichment analyses reveal the activation of functional pathways for DEGs in Ehmt2fl/fl animals compared to their respective Ehmt2+/+ counterparts. (D) QuanTIseq deconvolution analysis predicts immune cell composition based on the percentage of total immune cells.
Next, we performed integrative analysis and functional modeling by using the natural language processing-based identification of gene sets relationships from the ShinyGO suite combined with semantic-based algorithms using the DEGs from the across the different transcriptional landscapes as input (Ge et al., 2020). This approach yielded several functional gene groups associated with pancreatic maturation in the Ehmt2fl/fl pancreas. For instance, we found higher representation of genes related to multiple diverse cellular functions including adhesion, extracellular matrix organization (ECM), inflammation response, stress, apoptosis, digestive enzymes, metabolism, transport, epigenetics, and cell motility (Table 1). We also noted a differential activation of the hemoglobin locus control region (LCR) for Hba-a1, Hba-a2, Hbb-bs, and Hbb-bt, which serves as a pathognomonic indicator of alterations in nuclear 3D organization upon Ehmt2 loss due the subsequent decrease of H3K9me2 domains associated with reorganization of the active compartment in the nucleus (Yan et al., 2020; Fukuda et al., 2021; Takase et al., 2023). Furthermore, transcription factor motif analysis identified key upstream regulators of all DEGs related to Ehmt2 inactivation, among which we find main transcriptional regulators of several pathways congruent with the mSigDB functional enrichments (enrichment of transcription factor binding motifs in DEG promoters; Table 2). In summary, loss of Ehmt2 results in dysregulation of the epigenetic landscape affecting essential signaling pathways, such as p53 and KRAS, which are known to cause replication stress, arrest cell proliferation, and induce apoptosis, combined with changes in the immune cell populations, suggesting an important function for Ehmt2 in maintaining pancreatic homeostasis and a susceptibility of Ehmt2fl/fl mice to heightened injury-inflammation repair responses.
Table 1. Functional annotation of DEGs regulated by Ehmt2 inactivation in young adult mice.
Table 2. Transcriptional factor enrichment of DEGs upon Ehmt2 inactivation in young adult mice.
3.2 Ehmt2 inactivation in acinar pancreatic cells enhances the response to organ inflammationTo evaluate the relative contribution of Ehmt2 to the epithelial cell response during pancreatic inflammation, we repeatedly injected Ehmt2+/+ and Ehmt2fl/fl mice with caerulein to induce acute pancreatitis and performed RNA-seq. PCA analysis showed a clear separation of mice subjected to caerulein compared to their untreated counterparts (Figure 2A). Notably, the untreated samples, regardless of Ehmt2 inactivation status, clustered closely together on one end of the PCA plot. In contrast, samples undergoing acute pancreatitis displayed a distinct separation between the Ehmt2+/+ and Ehmt2fl/fl pancreatic samples (Figure 2A). Using pairwise analysis between caerulein and untreated groups, we found 5,263 DEGs. Of these DEGs, 1,812 were shared between Ehmt2+/+ and Ehmt2fl/fl with acute pancreatitis, consisting of 764 upregulated and 1,048 downregulated DEGs (Figures 2B, C; Supplementary Table S2). In terms of unique DEGs, 1,838 were distinctly upregulated upon acute inflammation in the Ehmt2fl/fl mice versus 189 in the Ehmt2+/+ group (Figure 2B). Correspondingly, 1,283 were exclusively downregulated during acute pancreatitis in the Ehmt2fl/fl animals compared to 142 in the Ehmt2+/+ (Figure 2C). We used RT-qPCR to validate a set of upregulated and downregulated gene targets among the most significant DEGs identified in Ehmt2fl/fl animals with acute pancreatitis (Supplementary Figure S2). Interestingly, a large subset of the genes that were found in the Ehmt2+/+ animals, which would constitute the response to acute pancreatitis, overlapped with the Ehmt2fl/fl mice (80% of upregulated and 91% of downregulated genes). Moreover, 70% of upregulated genes and 55% of downregulated genes that were differentially regulated in Ehmt2fl/fl animals were unique to the loss of Ehmt2. This highlights that Ehmt2 plays a major role during acute pancreatitis in maintaining transcriptional homeostasis in response to injury. Through heatmap clustering analyses of these DEGs, we identified that the genes differentially regulated in both Ehmt2+/+ and Ehmt2fl/fl animals presented with an amplified response when Ehmt2 was inactivated (Figure 2D). This finding in the Ehmt2fl/fl mice with acute pancreatitis is congruent with a principal role of Ehmt2 in mediating gene expression in response to tissue injury. In our analysis of the upregulated and downregulated genes, we found that Ehmt2+/+ and Ehmt2fl/fl mice with acute pancreatitis exhibited significant enrichment of similar pathways, specifically genes involved in injury-inflammation through activation of the oncogenic KRAS, P53, TNFα and TGF-β signaling pathways (Figure 2E). To delve deeper, we manually examined genes upregulated in acute pancreatitis compared to untreated mice, focusing on the subset that were uniquely activated or further de-repressed during the inflammatory response upon Ehmt2 inactivation. This analysis revealed a network of upstream regulatory genes that include Il1b, Il1r1, Tnf, Csf3r and members of the Ccl family (summarized in Table 3), which are known recruit and activate immune cells. With Il1r1 serving as the primary receptor for Il1b, this pathway is recognized for its role in triggering NF-κB signaling pathway activation, which we also found. We also detected increased gene expression networks related to immune cell activation and function, such as Slamf and cell surface molecules receptors. Similarly, the upregulation of chemokine signaling by C-X-C motif chemokine ligand families, indicates a heightened recruitment and migration of immune cells to inflamed pancreatic tissue. Notably, we also found upregulation of integrin and Interleukin cytokines, with roles in immunomodulatory processes that influence the balance between pro-inflammatory and anti-inflammatory responses. Thus, the increased expression of various inflammatory mediators highlights a cascade of cellular events that seem to culminate in the initiation and propagation of the enhanced immune response discovered in the Ehmt2fl/fl mice. In addition, there was increased representation of genes involved in cytoskeleton and cell adhesion, transcriptional regulation, DNA replication and repair, metabolic and oxidative stress, and RNA processing that collectively function to enhance cell survival, proliferation, and tissue regeneration during pancreatitis (Table 3). Contrastingly, the downregulated genes showed substantial enrichment of metabolic and proteostasis pathways, such as fatty and bile acid metabolism and unfolded protein response. This observation highlights that acute pancreatitis causes drastic changes to the pancreas gene expression landscape, reflecting that normal functionality of the pancreas is reduced (Figure 2E). Furthermore, we observed an increased enrichment of genes involved in a repair response, such as activation of mTORC1, PI3K-Akt, androgen and estrogen hormone signaling pathways. Lastly, we observed activation of genes involved in other signaling networks to help the pancreas tissue cope with damage caused by acute pancreatitis, such as angiogenesis to compensate for increased metabolic demand (Figure 2E). Notably, in the Ehmt2fl/fl pancreas tissues, the enrichment and overall number of genes for each ontology was found to be of greater significance with an increased number of genes in each of the categories, specifically for the UV response, cholesterol, mTORC1, myogenesis, estrogen, and reactive oxygen related pathways (Figure 2E). Thus, this data further highlights the Ehmt2-mediated functions that are necessary for the injury-inflammation-repair response during acute pancreatitis.
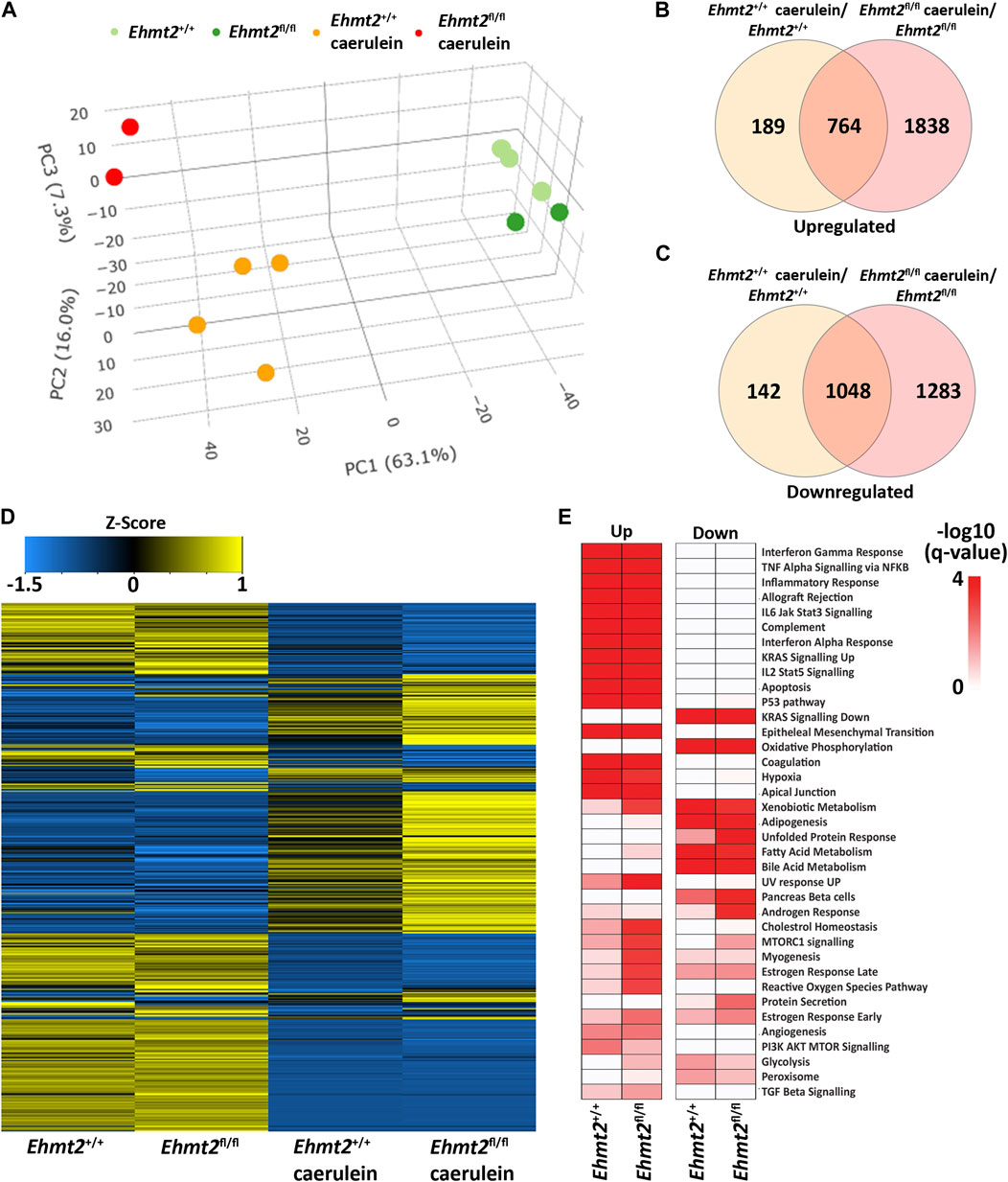
Figure 2. Ehmt2 is crucial for maintaining faithful transcriptional homeostasis in response to injury during acute pancreatitis. (A) PCA based on DEGs from RNA-seq conducted on pancreas tissue from Ehmt2+/+ and Ehmt2fl/fl adult mice both with and without induction of acute pancreatitis is shown. Venn diagram illustrates the overlap in significant DEGs between mice treated with caerulein and untreated mice, comparing Ehmt2+/+ and Ehmt2fl/fl animals for both (B) upregulated and (C) downregulated genes. (D) Heatmap displays the average expression of DEGs that were significant in at least one condition for Ehmt2+/+ and Ehmt2fl/fl animals in absence or presence of caerulein treatment. (E) MSigDB Hallmarks pathway enrichment analysis of significant DEGs between mice treated with caerulein and untreated mice in both Ehmt2+/+ and Ehmt2fl/fl animals is shown for upregulated and downregulated genes.
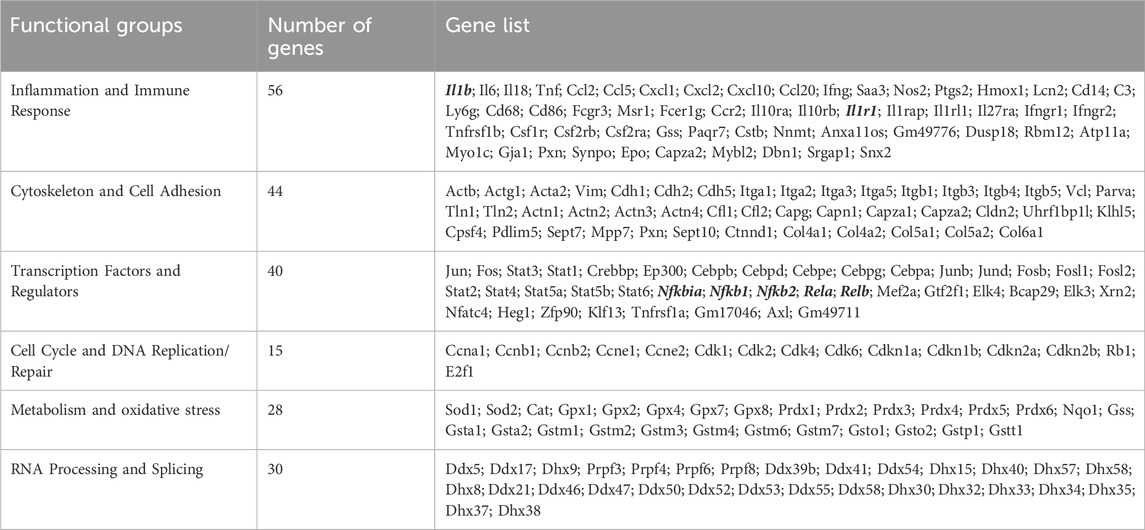
Table 3. Enhanced expression/de-repression of Ehmt2-regulated genes that are associated with the inflammatory and immune response in acute pancreatitis.
To reveal differences in the composition of the immune cell infiltrate across the total cells found between the Ehmt2+/+ and Ehmt2fl/fl pancreas of caerulein-treated animals, we used MCP Counter, an expanded deconvolution method due to the increased inflammation under these conditions (Figure 3A). The induction of acute pancreatitis highlighted immune cell signaling patterns that distinguished Ehmt2+/+ from Ehmt2fl/fl, also revealing an enhanced immune response when Ehmt2 function is lost with the number of markers for immune cells increasing by a total of 16.6%. Specifically, we identified an increased signal of 1.9% for T-cells in animals with Ehmt2 inactivation. Similar increases were found for canonical markers of neutrophils and myeloid dendritic cells with an increase of 1.9% and 1.2%, respectfully, for Ehmt2fl/fl animals compared to Ehmt2+/+ mice. Notably, macrophages represented the most substantial increase of 11.6% upon Ehmt2 knockout. (Figure 3A). These findings highlight the important role of Ehmt2 in regulating immune cell infiltration during acute pancreatitis. Thus, we used spatial transcriptomics to validate our findings from bulk RNA-seq. Spatial transcriptomics technologies, while unable to achieve single-cell resolution due to capturing gene expression data from regions containing on average 5-10 cells each, offer valuable insights by maintaining the spatial context within tissue samples (Grisanti Canozo et al., 2022). Analytically, we applied Seurat’s functionalities to define spatially variable genes (SVG) across tissue samples. To deconvolute the cell types within the spatial transcriptomic spots, we employed marker genes obtained from Zhou et al. (2022) and CellMarker (Zhang et al., 2019). We calculated the average number of spots containing SVGs indicative of immune infiltration by T cells (Cd3e), neutrophils (S100a8), dendritic cells (Ccr7), macrophages (Apoe), and NK cells (Nk7g) for Ehmt2+/+ and Ehmt2fl/fl mice. In concordance with our bulk RNA-seq data, we detected the enhanced immune response in Ehmt2fl/fl compared to the Ehmt2+/+ mice with comparable distributions of specific immune subtypes (Figure 3A). An average of 7,865 spots for Ehmt2+/+ and 7,542 for Ehmt2fl/fl mice were assigned in total across the tissue, and from these total spots, 0.8% contained SVG expression indicative of T-cell infiltrate in the Ehmt2+/+ pancreas, which increased to 1.9% in Ehmt2fl/fl. Similarly, we found higher proportions of markers for neutrophils and dendritic cells in Ehmt2fl/fl tissue (2.6% and 2.0%, respectively) compared to Ehmt2+/+ (0.5% and 0.6%, respectively). Once again, the macrophage population signified the most striking change between Ehmt2+/+ tissue (10.0%) and Ehmt2fl/fl (27.4%). Conversely, the difference in the proportions of NK cells were modest with 0.3% in Ehmt2+/+ to 0.8% in Ehmt2fl/fl (Figure 3A). To more comprehensively address the complex pathology of pancreatic disease, we expanded our spatial transcriptomic analysis to other relevant cell types using previously reported markers as labeled in Figure 3B (Olaniru et al., 2023). We found a significant reduction in spots associated with the SVG marker for acinar cells (Prss2) in Ehmt2fl/fl samples, with only 66.6% of spots indicative of harboring acinar cells (Figure 3B). In contrast, Ehmt2+/+ tissues displayed a more typical distribution, with 94.4% of spots representing the presence of acinar cells. Similarly, Ehmt2fl/fl exhibited a decrease in duct cells compared to Ehmt2+/+ (39.1% vs. 30.0%). On the other hand, features for islets (Ins2) (15.9% vs. 17.4%), fibroblasts (Cola1a) (10.6% vs. 12.7%), stellate cells (Mmp14) (1.1% vs. 2.3%), and tuft cells (Dclk1) (1.0% vs. 2.2%) showed slight increases when comparing Ehmt2+/+ tissues to Ehmt2fl/fl. Lastly, we evaluated SVGs that are hallmarks of epithelial-mesenchymal transition (EMT), given that this process is closely linked to pro-fibrotic signaling and activation of pancreatic stellate cells (Tian et al., 2016). Focusing on the gene Spp1, we found 25.7% of spots positive for EMT-like SVG expression in Ehmt2+/+ mice with acute pancreatitis, which notably increased to 45.7% in Ehmt2fl/fl pancreas (Figure 3B). As the macrophage marker showed the highest increase across the immune cells, we correspondingly found a clear increase of high expressing Apoe spots, suggesting a higher infiltrate and immune response when Ehmt2 is inactivated (Figure 3C). Thus, the results from spatial transcriptomics fundamentally confirmed our findings in bulk RNA-seq that Ehmt2 loss in the acinar cell triggers an increased immune infiltrate upon induction of acute pancreatitis.
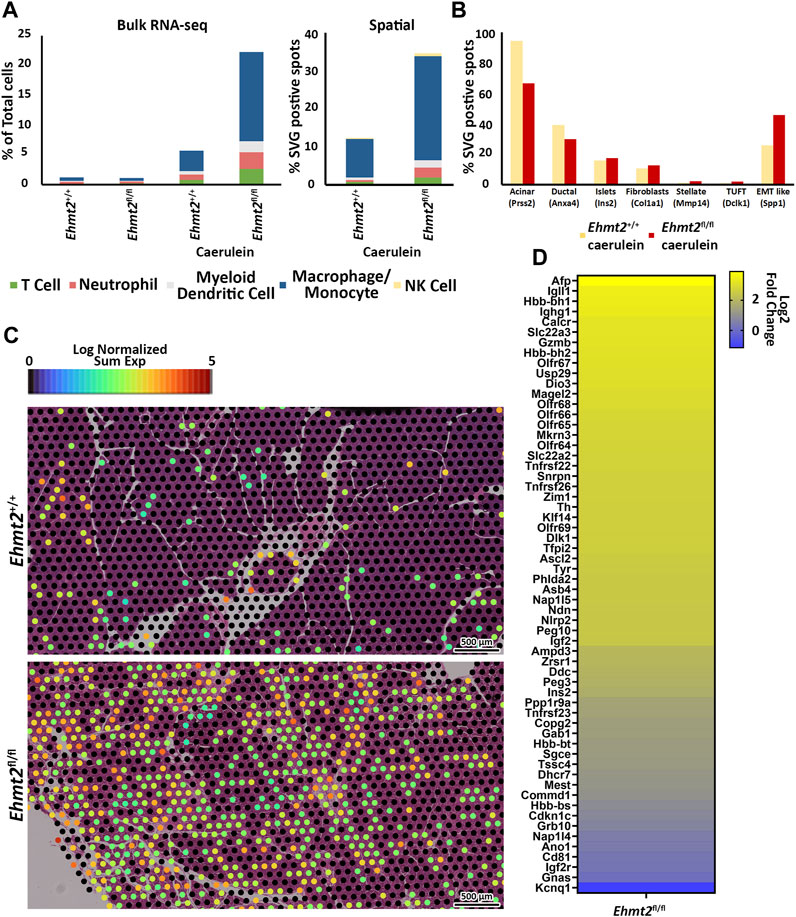
Figure 3. Ehmt2 deficiency promotes immune cell infiltration in injured pancreatic tissue. (A) MCPcounter deconvolution analysis predicts immune cell composition in the total cell population using Bulk RNA-seq data from Ehmt2+/+ and Ehmt2fl/fl animals, in the absence or presence of caerulein treatment. Manual spatial deconvolution is conducted using spots positive for SVGs and stringent immune cell markers in pancreas samples from caerulein-treated Ehmt2+/+ and Ehmt2fl/fl animals. (B) Bar graph shows pancreas cell deconvolution utilizing spots positive for SVGs of markers as indicated for a subset of pancreatic cells in pancreas tissue from caerulein-treated Ehmt2+/+ and Ehmt2fl/fl animals. (C) Visualization of macrophage SVG-positive spots is overlayed on caerulein-treated Ehmt2+/+ and Ehmt2fl/fl pancreas samples. (D) Heatmap displays log2 fold change for 60 genes found regulated by locus control regions and imprinted control regions.
Subsequently, we compiled a well-categorized list of genes known to be regulated by chromatin structure and nuclear organization, including several LCR and Imprinted Control Region (ICR) genes and evaluated the log2 FC by spatial transcriptomic analysis, comparing Ehmt2fl/fl to Ehmt2+/+ mice (Supplementary Table S3) (Moon and Ley, 1990; May and Enver, 1995; Terajima et al., 1995; Johnson et al., 1999; Yui et al., 2001; Li et al., 2002; Kajiyama et al., 2006; Yang and Corces, 2011; Thakur et al., 2016). This analysis revealed significant dysregulation of 60 LCR/ICR-controlled genes, specifically for LCRs such as Afp, Gzmb, and Hbb, and several imprinted genes in Ehmt2fl/fl mice. Notably, the well-studied Chr7 ICR1 and ICR2, known to control allelic expression of several genes in that region such as Kcnq1, Cdkn1c, H19, and Igf2, exhibited complete loss of preferential allelic expression, with both maternal and paternal genes showing derepression, except for Cd81 and Kcnq1, suggesting disruption of chromatin structure and nuclear organization (Figure 3D). This looping mechanism plays a crucial role in controlling gene expression by altering active and inactive compartments of topologically associating domains in 3D. These collective findings suggest that Ehmt2 helps preserve the epithelial characteristics of acinar and ductal cells in response to pancreatic injury, particularly during instances such as acute pancreatitis. This involvement is underscored by its role in gene repression and the regulation of 3D nuclear localization, emphasizing the multifaceted mechanisms through which Ehmt2 contributes to the maintenance of cellular identity in the context of physiopathological stimuli.
3.3 Ehmt2-mediated pathways during pancreas inflammation are model-independentTo confirm the enhanced tissue damage response during acute pancreatitis observed with loss of Ehmt2, we performed a pairwise analysis of the pancreatitis transcriptional landscape of the Ehmt2 knockout generated either with the Pdx1-Cre (Pdx1-Ehmt2fl/fl) or P48Cre/+ (P48-Ehmt2fl/fl) -driven models. This analysis aimed to identify genes involved in the injury response during acute pancreatitis that are transcriptionally regulated by Ehmt2, independent of the model used. PCA demonstrated that animals with intact Ehmt2 clustered together during the acute pancreatitis treatment regardless of the model, Pdx1 or P48Cre/+, and separated from their Ehmt2fl/fl counterparts (Figure 4A). Analysis of both models together revealed 775 DEGs with the P48-Ehmt2fl/fl and 954 DEGs with the Pdx1-Ehmt2fl/fl model compared to their Ehmt2+/+ counterparts (Figures 4B, C). Across DEGs, 486 genes were shared among Ehmt2fl/fl mice with acute pancreatitis, regardless of the Cre-driver used (Figures 4B, C; Supplementary Table S4). Specifically, we found 650 upregulated DEGs in the Pdx1-Ehmt2fl/fl and 518 with the P48-Ehmt2fl/fl, of which 347 DEGs were the same between the two models (Figure 4B). For downregulated DEGs, there were 139 DEGs in common between both models from the 304 downregulated DEGs in Pdx1-Ehmt2fl/fl and 257 in the P48-Ehmt2fl/fl tissues (Figure 4C). The reproducibility and distinct separation between acute pancreatitis-induced animals with Ehmt2 intact and those with Ehmt2 loss, regardless of the Cre model, present in the PCA (Figure 4A) are also evident in the heatmap (Figure 4D). Ontological analysis of DEGs revealed significant enrichment of distinct functional groups, including inflammatory response and immune signaling, cell signaling and proliferation, as well as metabolism and hormonal regulation. For instance, upregulated DEGs in both the Pdx1-Ehmt2fl/fl and P48-Ehmt2fl/fl acute pancreatitis groups, that were classified within the inflammatory response and immune signaling functional group, were enriched in pathways such as TNFα signaling via NF-kB, interferon gamma response, allograft rejection, IL-6/JAK/STAT3, and IL-2/STAT5 signaling. For the cell signaling and proliferation functional group, we found enrichment in KRAS signaling up, EMT, angiogenesis, the p53 pathway, and apoptosis, cholesterol homeostasis, xenobiotic metabolism, estrogen response late, and estrogen response early were enriched under metabolism and hormonal regulation DEGs for the Pdx1-Ehmt2fl/fl mice. Similar enrichment was observed for upregulated DEGs in P48-Ehmt2fl/fl mice. Conversely, downregulated DEGs in the Pdx1-Ehmt2fl/fl and P48-Ehmt2fl/fl acute pancreatitis models were only significantly enriched for networks that signal KRAS down (Figure 4E). Moreover, we performed MCP Counter deconvolution to evaluate the immune cell infiltrate of the P48-Ehmt2+/+ and P48-Ehmt2fl/fl animals with acute pancreatitis (Supplementary Figure S3A), finding similar changes with Ehmt2 knockout as the Pdx1-Cre acute pancreatitis model (Figure 3A), namely, an increase in markers for macrophages, T-cells, and neutrophils. To further investigate the role of Ehmt2 in regulating key transcriptomic responses in pancreatitis, we concentrated on the 486 DEGs consistent across both models. We considered these DEGs as core genes regulated by Ehmt2 and used ChIP-Atlas, a comprehensive data-mining suite for exploring epigenomic landscapes, to access publicly available datasets (Zou et al., 2022). From this resource, we identified 15,851 target genes associated with Ehmt2 in both human and mouse genomes. Notably, 266 of the 486 DEGs (55%) have been recognized as bona fide targets of Ehmt2 (Supplementary Table S5). Moreover, pathway enrichment analysis of this subset of 266 confirmed Ehmt2 targets closely mirrors the significant enrichment observed in the core genes regulated by Ehmt2, including pathways involved in cell signaling and proliferation, EMT, and inflammation (Supplementary Figure S3B). Thus, whether Ehmt2 is deleted from the pancreas using either the Pdx1-Cre or P48Cre/+ model, the modified transcriptional impact of acute pancreatitis compared to their counterparts with Ehmt2 intact are similar, highlighting that Ehmt2 participates primarily in preventing an enhanced injury-inflammatory repair response in this organ.
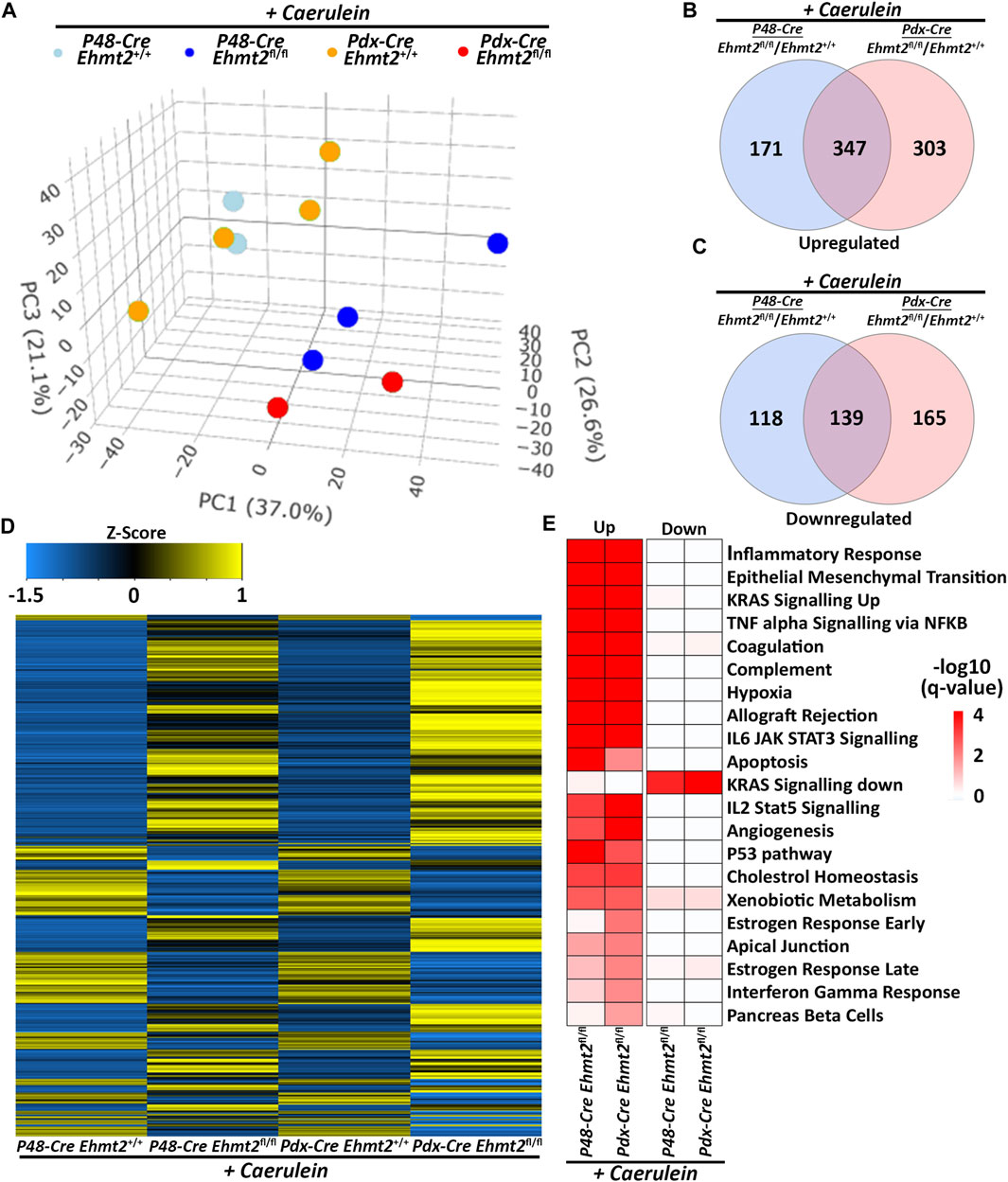
Figure 4. Ehmt2-mediated transcriptional regulation during acute pancreatitis is model independent. (A) PCA based on DEGs from RNA-seq conducted on pancreas tissue from adult Ehmt2+/+ and Ehmt2fl/fl mice with acute pancreatitis for both Pdx1-Cre and P48-Cre driven models is shown. Venn diagram illustrates the overlap in significant DEGs between pancreatitis-induced Pdx1-Cre and P48-Cre models with Ehmt2 inactivation compared to their Ehmt2+/+ counterparts for (B) upregulated and (C) downregulated genes. (D) Heatmap displays the average expression of DEGs that were significant in at least one condition for Ehmt2+/+ and Ehmt2fl/fl animals treated with caerulein in both Pdx1-Cre and P48-Cre models. (E) MSigDB Hallmarks pathway enrichment analysis is shown for significant DEGs between caerulein-treated Pdx1-Cre and P48-Cre models in both Ehmt2+/+ and Ehmt2fl/fl animals for both upregulated and downregulated genes.
Lastly, we performed further analysis using NLP and semantic-based algorithms on DEGs identified by the Pdx1 and P48 models to determine shared pathways, which included signal transduction and immune defense (Table 4). We found enrichment in cell adhesion and extracellular matrix (ECM) genes, indicating changes in ECM organization, tissue remodeling processes, and immune cell trafficking within the pancreas during pancreatitis. Moreover, Ehmt2fl/fl animals upregulated several enzymes, transporters, and channels, which likely reflects the increased metabolic needs (Table 4). Additionally, we noted that derepression of Il1b and its known receptor Il1r1, as well as increased expression of upstream regulators involved in the NF-κB immune-mediated response were present in both the Pdx1 and P48 models. Analysis of enriched cis-regulatory elements within promoter regions of these DEGs revealed associations with universal stripe factors such as ZFP281, PATZ1, SP, and KLF5 among others (Zhao et al., 2022). This suggests that Ehmt2 may play a role in the crosstalk mechanism between these universal stripe factors, facilitating chromatin accessibility (Table 5). Thus, the response to inflammation that we describe here for loss of Ehmt2 in the pancreas epithelial cell is primarily not mediated by tissue enriched Stripe Factors but rather general Stripe factors that drive responses in many organs, suggesting that these processes may have broader relevance.
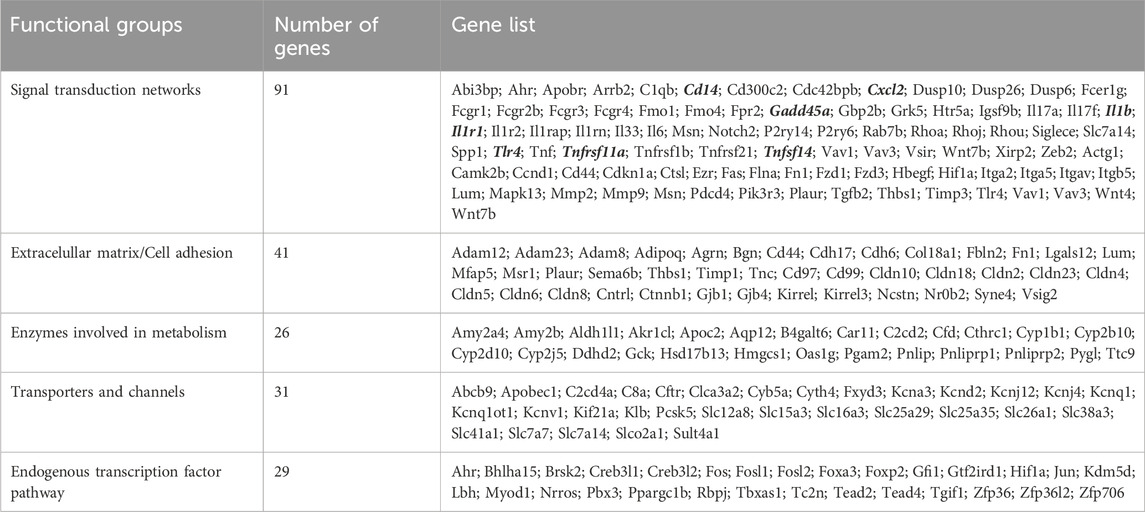
Table 4. Integrative analysis by biological modeling of model-independent DEGs to determine shared Ehmt2-dependent pathways.
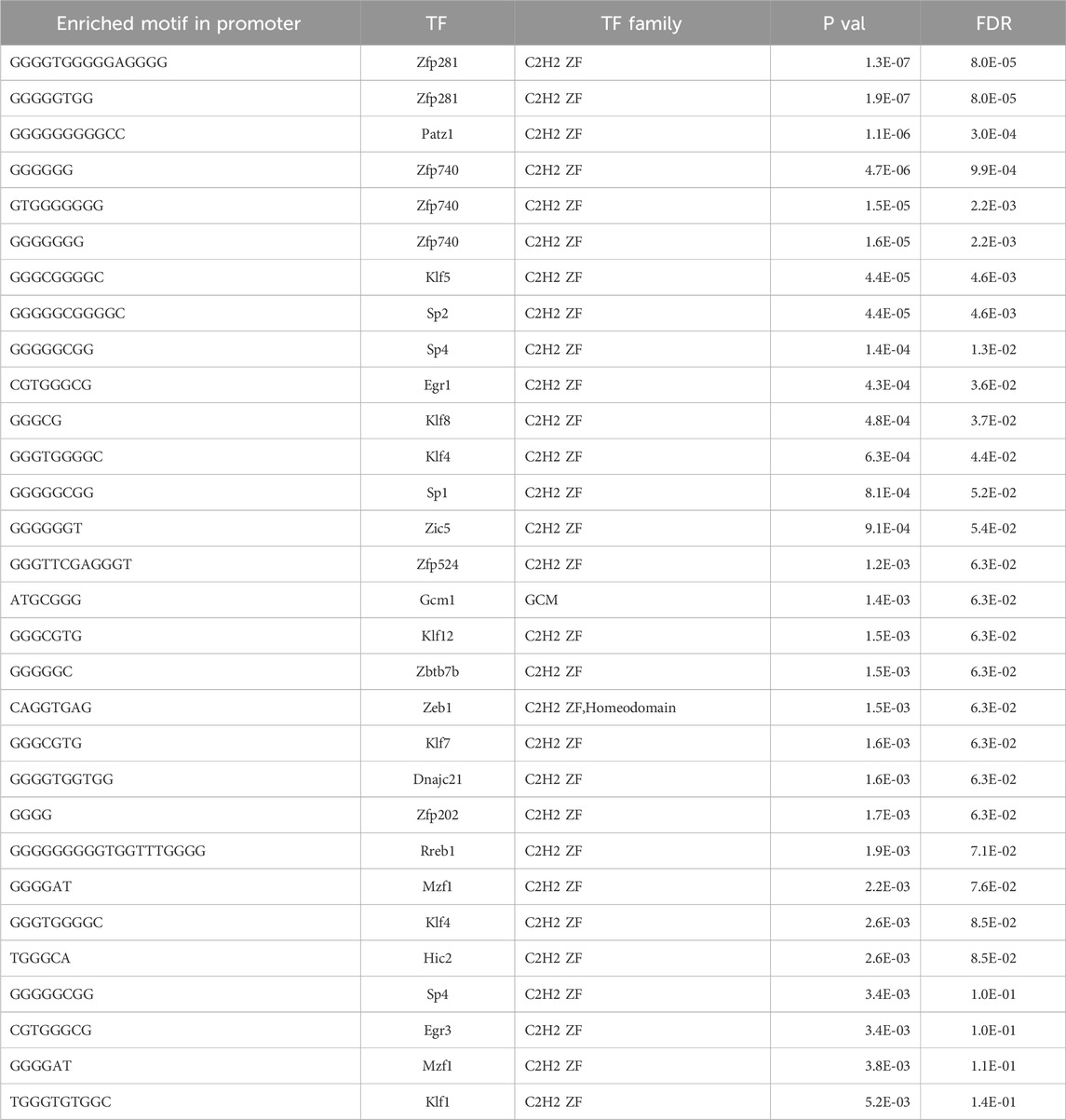
Table 5. Transcriptional factor enrichment of DEGs by Ehmt2 during acute pancreatitis.
3.4 Morphological and biochemical assessment confirms the impact of Ehmt2 inactivation on pancreas inflammation in both the Pdx1-and P48-driven modelsAcute pancreatitis in humans is linked to both local and systemic complications. Systemic complications typically present as organ failure, observed in about 20% of cases (Garg and Singh, 2019). Thus, biochemical investigation was performed on serum from both Pdx1-Cre and P48Cre/+ caerulein-treated mice with or without Ehmt2 to evaluate liver and kidney function. We found significantly decreased levels of albumin, total protein, blood urea nitrogen (BUN) and alkaline phosphatase (ALP) levels in Ehmt2fl/fl animals compared to Ehmt2+/+ mice (Figure 5A). The decrease in circulating albumin, coupled with heightened inflammation in Ehmt2fl/fl mice, suggests a potential increase in capillary permeability and the efflux of circulating albumin into interstitial spaces (Soeters et al., 2019), which is seen in cases of acute pancr
留言 (0)