Study design
This was a two-period, open-label, single-dose phase 1 study (NCT03811834) conducted at one clinical site (Celerion, Lincoln, Nebraska, USA) in healthy male participants. The primary objective of Period 1 was to determine the absolute bioavailability of mobocertinib following a single oral dose of 160 mg mobocertinib as capsules and a single intravenous (IV) microdose of 50 µg (~ 2 µCi) [14C]-mobocertinib. The primary objectives of Period 2 were to (1) assess the cumulative excretion of total radioactivity in urine and feces (mass balance); (2) assess the metabolite profile of mobocertinib in plasma, urine, and feces; and (3) characterize the PK of mobocertinib, AP32960, and AP32914 in plasma, whole blood, and urine, and total radioactivity concentration equivalents in plasma and whole blood following a single oral dose of 160 mg (~ 100 µCi) [14C]-mobocertinib as an oral solution. A sample size of 6 healthy males was selected without statistical considerations and deemed adequate to meet the study objectives. Furthermore, the sample size was limited based on clinical considerations for human ADME studies and to limit exposure to radioactivity.
Period 1: Absolute Bioavailability Study
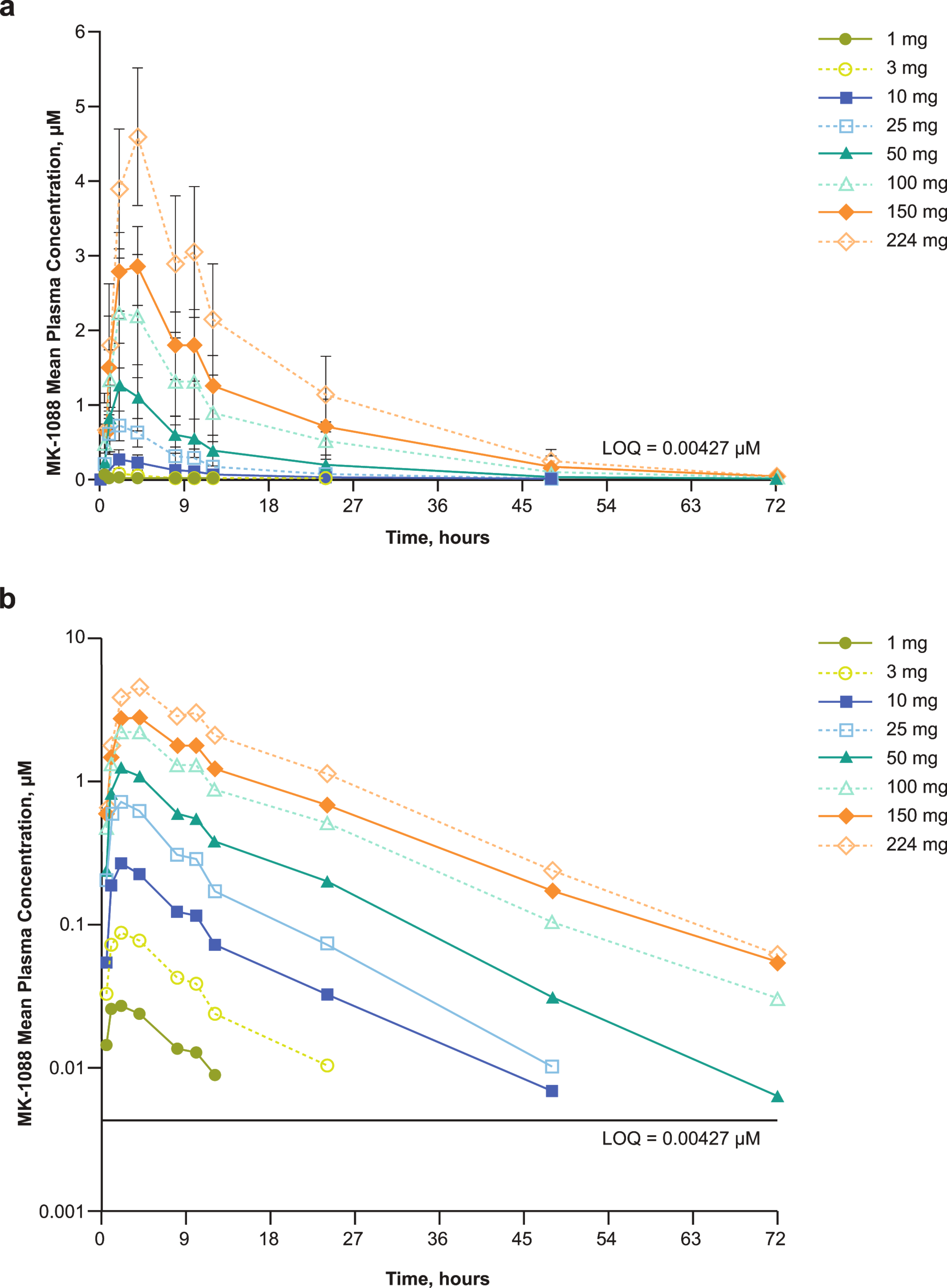
On Day 1 of Period 1, following an overnight fast of at least 10 h, participants received a single 160 mg dose of non-radiolabeled mobocertinib as capsules. At 3.75 h post oral dosing (i.e., 15 min before the median oral tmax of ~ 4 h), participants received a nominal dose of 50 µg (~ 2 µCi) [14C]-mobocertinib as a 15-minute IV infusion. However, due to nonspecific binding of [14C]-mobocertinib to the dosing syringe and tubing, actual doses administered ranged from 36.8 to 38.7 µg (1.47 to 1.55 µCi). Participants were required to stay in the clinic from Day − 1 through at least the 96-hour blood draw (Day 5) or until a discharge criterion was met (i.e., ≥ 80% of the total administered radioactive dose was recovered in urine and fecal samples or excretion of radioactivity in urine and feces combined had declined to ≤ 1% of the total administered dose per day for ≥ 2 consecutive intervals), up to a maximum of 7 days postdose (Day 8). Blood samples for the measurement of plasma mobocertinib, AP32960, and AP32914 concentrations were collected predose and at 0.5, 1, 2, 3, 4, 5, 6, 8, 12, 24, 36, 48, 72, and 96 h after oral administration. Additional blood samples to determine [14C]-total radioactivity, [14C]-mobocertinib, [14C]-AP32960, and [14C]-AP32914 in plasma were collected at 3.75 h after oral dosing (i.e., predose for [14C] assessments), at the end of the IV infusion, and at 10, 20, and 30 min, and 1, 2, 4, 8, 20, 32, 44, 68, and 92 h after the end of infusion. For participants who did not meet the discharge criteria by Day 5, blood samples continued to be collected in 24-hour intervals until a discharge criterion was met or up to Day 8. Urine and feces were collected before oral dosing (− 48–0 h); urine was collected over 0–3.75 h, 3.75–12 h and 12–24 h after oral dosing; feces were collected from 0 to 3.75 h and 3.75–24 h after oral dosing; both urine and feces were then collected over 24-hour intervals until a discharge criterion was met, or up to Day 8 (168 h postdose).
Period 2: Human ADME Study
After a washout of 8–9 days, participants returned to the clinic for Day − 1 of Period 2. On Day 1 of Period 2, after a fast of ≥ 10 h, participants received a single nominal dose of 160 mg (~ 100 µCi; whole body effective dose of 5.3 mrem for a 70-kg male human) [14C]-mobocertinib as a 70 mL oral solution. The actual doses administered ranged from 161 to 163 mg (92.4 to 93.9 µCi). Participants stayed in the clinic from Day − 1 until a discharge criterion (≥ 80% of the total administered radioactive dose was recovered in urine and feces or excretion of radioactivity in urine and feces combined had declined to ≤ 1% of the total administered dose per day for ≥ 2 consecutive intervals) was met, or up to 10 days postdose. To measure the total radioactivity and concentrations of mobocertinib, AP32960, and AP32914 in whole blood and plasma, 4 blood samples were collected predose, and at 0.5, 1, 2, 3, 4, 5, 6, 8, 12, 24, 36, 48, 72, 96, 120, 144, 168, 192, 216, and 240 h postdose; a fifth blood sample was collected for plasma metabolite profiling at predose and 1, 2, 4, 6, 12, 24, 48, 72, 96, 120, and 168 h postdose. Feces and urine were collected predose (feces within 48 h and urine within 24 h prior to dosing), 0–24 h (urine from 0–12 and 12–24 h), and then at 24-hour intervals up to 240 h postdose or until a discharge criterion was met. Two participants did not meet a discharge criterion by Day 11. These 2 participants continued with at-home fecal sample collections. One participant provided fecal samples until Day 13 (264–288 h interval) and one participant provided fecal samples until Day 19 (408–432 h interval), after which they met a discharge criterion.
All participants were contacted 30 days after the last dose of study drug for safety follow-up.
Participants
Eligible participants were healthy, adult, male non-smokers, aged 19–55 years with a body mass index ≥18.0 and < 30.0 kg/m2 and medically healthy with no clinically significant medical history, physical examination, laboratory profile, vital sign, or electrocardiogram (ECG) findings. Key exclusion criteria were QT interval corrected for heart rate using Fridericia’s formula (QTcF) interval > 460 ms; estimated creatinine clearance < 80 mL/min; infrequent bowel movements within the previous 30 days or recent history of abnormal bowel movements (e.g., diarrhea, constipation) within 2 weeks before first dose; inability to refrain from use of any prescription or non-prescription medication, herbal remedy, or vitamin supplement within 2 weeks before first dose; and use of inducers of CYP3A and/or P-glycoprotein within 28 days before first dose and throughout the study. Full eligibility criteria are provided in Supplementary Table S1.
Bioanalytical methods
Plasma, whole blood, and urine samples were assayed for mobocertinib, AP32960, and AP32914 concentrations using liquid chromatography-tandem mass spectrometry (Q2 Solutions, Ithaca, New York, USA). The analytical range for each analyte was 0.250 to 500 ng/mL in whole blood and plasma, and 1.00 to 1000 ng/mL in urine. Plasma, urine, and fecal samples from Period 1 were assayed for [14C]-mobocertinib, [14C]-AP32960, and [14C]-AP32914 concentrations, and plasma samples were assayed for total radioactivity concentration equivalents using accelerator mass spectrometry (AMS; Pharmaron, Germantown, Maryland, USA), with lower limits of quantitation (LLOQs) for each analyte of 1.20 pg/mL in plasma and 6.99 pg/mL in urine and feces; the LLOQ for total radioactivity concentration equivalents in plasma ranged from 0.972 to 2.00 pg eq/mL across participants. Urine and fecal samples were assayed for total radioactivity following the IV dose of [14C]-mobocertinib in Period 1, and whole blood, plasma, urine, and fecal samples were assayed for total radioactivity following the oral dose of [14C]-mobocertinib in Period 2 using liquid scintillation counting (LSC; Celerion, Lincoln, Nebraska, USA; LLOQs: Period 1 urine: 0.115–0.118 ng eq/g, feces: 0.720–0.759 ng eq/g; Period 2 whole blood: 87.3–112 ng eq/g, plasma: 116–132 ng eq/mL, urine: 37.8–46.8 ng eq/g, feces: 110–336 ng eq/g across participants). Urine and fecal samples collected in Period 1 that had total radioactivity concentration equivalents below the LLOQ of the Celerion LSC assay were sent to Pharmaron for measurement by AMS (LLOQ: urine: 0.0000253–0.00111 ng eq/g; feces: 0.000404–0.0278 ng eq/g across participants). The LSC and AMS assays were not cross-validated.
Pharmacokinetic analyses
Noncompartmental PK parameters were calculated for analytes based on total concentrations in whole blood and plasma using Phoenix WinNonlin versions 7.0 and 8.1 (Certara, Princeton, New Jersey, USA). Derived PK parameters included tmax, maximum observed concentration (Cmax), AUC from time 0 to the last quantifiable concentration (AUClast), AUC from time 0 to infinity (AUC∞), apparent clearance after oral administration (CL/F; mobocertinib only), and apparent volume of distribution during the terminal disposition phase after oral administration (Vz/F; mobocertinib only). In Period 1, mobocertinib clearance (CL) and volume of distribution during the terminal disposition phase (Vz) were calculated after IV administration. Blood-to-plasma ratios for Cmax and AUC∞ were calculated for mobocertinib, AP32960, and AP32914 in Period 2. The absolute bioavailability of mobocertinib was estimated by comparing the ln-transformed AUC∞ of mobocertinib in plasma following the single oral dose of mobocertinib 160 mg as capsules and the ln-transformed dose-normalized (to 160 mg) AUC∞ of [14C]-mobocertinib following the single IV microdose in Period 1 using an analysis of variance model with route of administration (oral/IV) as a fixed effect and participant as a random effect. The geometric mean ratio and 90% CIs were expressed as a percentage relative to IV administration.
PK parameters for analytes in urine and feces were calculated using SAS Version 9.4 and included the cumulative amounts and percentages of the administered radioactive dose excreted as [14C]-mobocertinib, [14C]-AP32960, and [14C]-AP32914 in urine and feces following IV dosing in Period 1 and oral dosing in Period 2. Renal clearance (CLR) was calculated as the cumulative amount of radiolabeled mobocertinib, AP32960, or AP32914 recovered in urine divided by the plasma AUC to the time of the last common time point at which an analyte was quantifiable in both urine and plasma for individual participants (AUCCLR) following oral dosing of [14C]-mobocertinib in Period 2. Mass balance was defined as the percentage of total radioactivity recovered in urine and feces combined relative to the total amount of the administered radioactive dose.
Safety assessments
Safety was evaluated based on the incidence and severity of AEs and changes from baseline in clinical laboratory results, vital signs, and ECG parameters. AEs were graded according to Common Terminology Criteria for Adverse Events version 5.0.





留言 (0)