
記住我

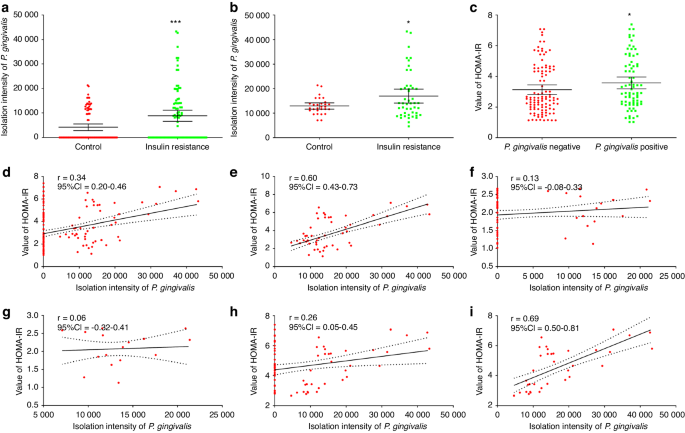


Human dental pulp stem cells (hDPSCs) isolated from the teeth of healthy volunteers are characterized by multi-differentiation potential and MSC-like surface markers. HDPSCs penetrated out from dental pulp tissue fragments and proliferated to form dense colonies that present typical fibroblastic morphology (Figure S1a, P0-P3). The multipotency of hDPSCs were identified by osteogenic, chondrogenic, and adipogenic differentiation. After staining with Alizarin Red, Alcian Blue, and Oil Red O, the results showed that the presentation of calcium phosphate, proteoglycans, and lipid droplets under the corresponding induced cues (Figure S1b). Flow cytometry results performed to evaluate the MSC-like properties of hDPSCs also showed that hDPSCs expressed CD44, CD73, and CD90 positively and CD34, CD45, and HLA-DR negatively (Figure S1c).
Prolonged periods of uncontrolled intracerebral homeostatic imbalances lead to abnormal activation of microglia and chronic neuroinflammation, which can further exacerbate neurodegeneration and cognitive impairment.1,14 To investigate the effects of hDPSCs on microglia abnormal activation pathology, we induced BV2 cells with bacterial lipopolysaccharide (LPS) stimulation, which is considered the gold standard for microglia activation.15 BV2 cells were challenged with different conditions of LPS to determine the best induced effects. As previously described,16,17 BV2 cells were incubated with LPS at a final concentration of 1ug/ml for 24 h, resulting in decreased cell viability and significantly increased expression of pro-inflammatory factors (Fig. 1a, b). Light microscope results of LPS-induced BV2 cells in vitro showed that the cell morphology of the LPS-induced BV2 cells exhibited a branching morphology and larger volumes compared with normal group; Co-cultured with hDPSCs reversed the stimulatory reactivity (Fig. 1c). In AD, microglia, which originally play a neuroprotective role by degrading Aβ plaques through phagocytosis, are abnormally activated into a pro-inflammatory state and release various neurotoxic molecules.15,18 We next investigated whether hDPSCs influenced microglia reactivity and homeostasis by measuring levels of inflammatory factors and phenotypic changes associated with morphological activation. We found a strong reduction in mRNA levels of pro-inflammatory cytokines (IL-1β, IL-6, and TNF-α) in microglia after co-culture with hDPSCs, whereas the mRNA expression of anti-inflammatory cytokines (IL-10) was enhanced (Fig. 1d). Similar to the positive efficacy of hDPSCs regulating inflammatory cytokines at the transcriptional level, western blot also confirmed that hDPSCs significantly reduced the protein expression of LPS-induced pro-inflammatory cytokines in microglia (Fig. 1e, f). Intriguingly, the mRNA expression of pro-inflammatory cytokines in the hDPSCs-treatment group decreased even more than that in the WT group, which may be closely related to the strong regulation of transcription by stem cells. Considering that LPS is thought to be a representative inducer of microglial polarization,19 in this co-culture model, we also assessed whether hDPSCs affected microglial polarization in inflammatory injury conditions. In line with this, the immunostaining for the M1-phenotype marker INOS was decreased and the M2-phenotype marker Arg1 was significantly increased in the hDPSCs-treatment group than that in LPS group (Fig. 1g, h), suggesting that hDPSCs reduced the number of hyperinflammation-induced activated microglia, promoting M1-reactive microglia tendencies toward the M2-protective phenotype. In the control group of BV2 cells, there was little evidence of INOS-positive cells. These results demonstrated that when faced with a hyperinflammatory challenge, hDPSCs could reverse microglial hyperreactive states and mitigate inflammation levels.
Fig. 1
hDPSCs mitigated LPS-triggered inflammation in vitro. a Primary BV2 cells were induced at different time by increasing doses of LPS (0–6 μg/ml), then cell viability was measured by CCK-8 assay. b The mRNA expression of pro-inflammatory cytokine (TNF-α, IL-1β, and IL-6) in LPS-induced BV2 cells for 24 h. c The representative morphological images of BV2 cells in Control group, LPS-induced group and hDPSCs co-culture group were observed under light microscope. Scale =100 μm. d The mRNA expression of TNF-α, IL-1β, IL-6, and IL-10 in BV2 cells of three group. e, f Representative images and quantification of western blotting showed the expression of inflammatory-related factors (TNF-α, IL-1β, IL-6, and IL-10) in different treated BV2 cells. g, h Representative immunostaining images and quantification of markers for activated microglia (INOS, green and Arg1, red). Scale = 50 μm (n = 3 per group; Values represented mean ± SD; ns indicates no significant, *P < 0.05, **P < 0.01, ***P < 0.001)
hDPSCs ameliorated LPS-induced oxidative stress and apoptosis in BV2 cells by activating Nrf2 via the AKT/GSK3β pathwayIt has been shown that microglia or macrophages exposed to inflammatory stimuli undergo metabolic reprogramming, facilitating cytokine and ROS production.20 Given the downregulation of the pro-inflammatory genes, we suspected that hDPSCs modulated the microglial phenotypic transition by blocking the ROS-triggered inflammatory signaling pathway. Differences in microglial reactivity were further supported by changes in ROS after LPS or hDPSCs treatment, noted by an increased number of DCF-positive cells in the LPS-induced BV2 cells, and hDPSCs predominantly inhibited ROS production (Fig. 2a). The DCF fluorescence quantitative analysis also confirmed the result (Fig. 2b).
Fig. 2
hDPSCs ameliorated LPS-induced oxidative stress and apoptosis in BV2 cells by activating Nrf2 via the AKT/GSK3β pathway. a, b The reactive oxygen species (ROS, green) level in BV2 cells detected by DCFH-DA staining and statistically analyzed. Scale = 100 μm. c, d Representative images and quantification of western blotting showed the expression of total Nrf2, HO-1, GPX4, SOD1, and nuclear Nrf2 in different treated BV2 cells. e Representative western blotting results showed the expression of p-AKT (ser473) and p-GSK3β (ser9) of BV2 cells in Con, LPS, and LPS+hDPSCs groups. f The quantification of p‐AKT (ser473) and p‐GSK3β (ser9), respectively, compared with total‐AKT and total‐GSK3β. g, h The apoptosis of BV2 cells in each group was determined by flow cytometry. i, j Representative images and quantification of western blotting showed the expression of BCL-2, Bax, and cleaved caspase 3 in different treated BV2 cells (n = 3 per group; Values represented mean ± SD; ns indicates no significant, *P < 0.05, **P < 0.01, ***P < 0.001)
Considering that we observed that hDPSCs significantly reduced LPS-induced neuroinflammation and ROS production in BV2 cells, we next hypothesized that hDPSCs might mediate the expression of relevant antioxidant enzymes, which are known as pivotal molecules in regulating oxidative stress and inflammation.21 RT-PCR assays manifested that consistent with foregoing experimental results, hDPSCs inhibited the decline in mRNA expression of the antioxidant factors Nrf2, HO-1, SOD1, and GPX4 (Figure S2a). Among them, Nrf2 acts as a major transcriptional regulator and translocates from the cytoplasmic to the nucleus to increase the activity of antioxidant response kinases, including H0-1, SOD, and GPX4. Meanwhile, the functional activation of Nrf2 is also controlled by upstream regulators phosphorylated protein kinase B (AKT) and phosphorylated glycogen synthase kinase 3β (GSK3β).8,22 Therefore, we further verified the protein expression of these genes. Western blotting revealed that hDPSCs significantly suppressed LPS-induced downregulation of Nrf2, HO-1, GPX4, and SOD1 by promoting the upregulation of p-AKT (ser473)/p-GSK3β (ser9) and the nuclear translocation of its downstream Nrf2 (Fig. 2c–f). High doses of LPS are well known to cause the production of inflammatory mediators and damaging molecules, but they also lead to toxic injury to the cell itself.19,23 Based on the AnnexinV-PI apoptosis experiment, we also verified that hDPSCs ameliorated LPS-induced BV2 cells damage because we observed a decrease in the percentage of apoptosis in the hDPSCs treatment group, although not as much as in the normal group (Fig. 2g, h). In line with this, further analyses found that in LPS-induced BV2 cells, co-culture with hDPSCs significantly increased the expression of the anti-apoptotic protein BCL2 and decreased the expression of apoptotic proteins cleaved caspase 3 and Bax (Fig. 2i, j).
To further confirm whether the AKT/GSK3β signaling pathway is critical for hDPSCs-induced nuclear accumulation of Nrf2, BV2 cells were preincubated with an AKT pathway inhibitor for 2 h, followed by hDPSCs treatment,24 and then the associated protein expression was detected. Western blot results showed that LY294002 significantly diminished hDPSCs-induced phosphorylation of p-AKT (ser473) and p-GSK3β (ser9) compared with hDPSCs-treated groups, which caused decreased protein expression of Nrf2 and its downstream gene HO-1 and reduced nuclear accumulation of Nrf2 (Figure S3a–d). Downregulation of LY294002-induced expression of antioxidant proteins also resisted the elimination of ROS by hDPSCs in LPS-induced BV2 cells (Figure S3e, f). Generally, these results demonstrated that hDPSCs can mitigate LPS-induced microglial damage by activating the AKT/GSK3β pathway, which enables the accumulation of Nrf2 in the nucleus to contain oxidative stress and neuroinflammation.
hDPSCs alleviated oxidative stress and mitochondrial damage in GLU-treated HT22 cellsPro-inflammatory M1 microglia not only produce pro-inflammatory factors and ROS but also enhance the excitotoxicity of glutamate, thus damaging neurons and the brain microenvironment.25 During the progression of AD, excessive extracellular glutamate concentrations stimulate glutamate receptors, which not only leads to intracellular calcium overload but also contributes to the production of ROS, resulting in cellular excitotoxicity and neuronal death.26,27 Therefore, based on the demonstrated ability of hDPSCs to shift microglia toward an anti-inflammatory phenotype and provide protection, we further examined whether hDPSCs have a neuroprotective effect in a glutamate-induced excitotoxicity in vitro AD model. As shown in Fig. 3a, b, CCK-8 and RT-PCR experiments were first performed to determine the optimal concentration of 6 mM/L glutamate to induce HT22 cells for 12 h. Using the co-culture models with hDPSCs and glutamate-induced HT22 (GLU group) (Fig. 3c), we observed the cell morphology by light microscopy and found that most of the neurons that lost their original cell morphology after being stimulated by glutamate gradually recovered normal cell morphology similar to that of the control group after co-culture with hDPSCs (Fig. 3d). Similar to the results of LPS-induced BV2 cells, HT22 cells exposed to glutamate clearly increased DCF-positive cells, while co-culture with hDPSCs obviously inhibited intracellular ROS production (Fig. 3e, f).
Fig. 3
hDPSCs alleviated oxidative stress and mitochondrial damage in GLU-treated HT22 cells. a Primary HT22 cells were induced at different time by increasing doses of GLU (0–10 μg/mL), then cell viability was measured by CCK-8 assay. b The mRNA expression of antioxidant cytokine (Nrf2, HO-1, GPX4, and SOD1) in GLU-induced HT22 cells for 24 h. c A transwell non-contact co-culture assay system was structured into the AD model. d The representative morphological images of HT22 cells in Control group, GLU-induced group, and hDPSCs co-culture group were observed under light microscope. Scale = 100 μm. e, f The reactive oxygen species (ROS, green) level in HT22 cells detected by DCFH-DA staining and statistically analyzed. Scale = 100 μm. g, h The mitochondrial ROS level in HT22 cells determined by MitoSOX red staining. Scale = 100 μm. i, j Mitochondrial membrane potential of the HT22 cells were detected by the JC-1 staining and the quantification of red/green fluorescence intensity. Scale bar = 50 μm. k Morphometric ultrastructural analyses by TEM showed the intracellular mitochondrial structure of HT22 in the three groups. Scale bar = 500 nm (n = 3 per group; values represented mean ± SD; ns indicates no significant, *P < 0.05, **P < 0.01, ***P < 0.001)
A closely association between physiological aging and declining mitochondrial function has long been noted.28 The genetic polymorphisms present in the etiology of a vast majority of patients with late-onset AD are also associated with age-dependent oxidative stress and mitochondrial abnormalities.29 Glutamate excitotoxicity induces an intracellular Ca2+ superload that impairs mitochondrial antioxidant activity and increases mitochondrial ROS production, ultimately leading to apoptosis.30,31 Next, we also tested whether hDPSCs also protect mitochondria. As evidenced by Mito-SOX red staining results, high concentrations of glutamate lead to ROS accumulation in the mitochondria of neuronal cells, and hDPSC significantly alleviated this characterization (Fig. 3g, h). Synchronously, to further evaluate the effect of hDPSCs on oxidative stress-induced mitochondrial damage, JC-1 staining, and transmission electron microscopy (TEM) were used to detect mitochondrial membrane potential (MMP) and structural integrity, respectively. As depicted in Fig. 3i, j, compared with GLU-induced HT22 cells, the depolarization of mitochondrial membrane potential was distinctly attenuated after hDPSCs treatment. Morphometric analyses with TEM indicated that the intracellular mitochondria of GLU-damaged HT22 cells presented swollen and vacuolated, whereas treatment with hDPSCs restored the mitochondria to a long shape resembling that of the WT group, and the mitochondrial cristae were also arranged neatly (Figs. 3k, S4). Taken together, these data illustrated that hDPSCs alleviated oxidative stress and mitochondrial damage in GLU-treated HT22 cells.
hDPSCs attenuated apoptosis in GLU-induced HT22 cells by activating Nrf2 via the AKT/GSK3β pathwayNext, considering the antioxidant activity of Nrf2 in BV2 cells, we conducted a more in-depth investigation of whether the antioxidant effect of hDPSCs is mediated by Nrf2 in GLU-induced HT22 cells. After co-culture of hDPSCs with GLU-treated HT22 cells, immunofluorescence staining and quantitative analysis showed that the overall Nrf2 expression level of neuronal cells treated with hDPSCs was higher than that of GLU group, and the accumulation/translocation of Nrf2 within the nucleus was significantly enhanced in hDPSCs-treated group (Fig. 4a, b). Similar results were verified by RT-PCR and western blot analysis, which showed that hDPSCs not only up-regulated the mRNA and total protein expression of Nrf2 and downstream antioxidant enzymes including HO-1, SOD1, and GPX4, but also increased the nuclear protein expression level of Nrf2 (Figs. 4c, d and S2b).
Fig. 4
hDPSCs attenuated apoptosis in GLU-induced HT22 cells by activating Nrf2 via the AKT/GSK3β pathway. a, b Representative immunofluorescent staining images and quantification of Nrf2 in three different groups. Scale = 20 μm. c, d Representative images and quantification of western blotting showing the expression of total Nrf2, HO-1, GPX4, SOD1, and nuclear Nrf2 in different treated HT22 cells. e Representative western blotting results showed the expression of p-AKT (ser473) and p-GSK3β (ser9) of HT22 cells in the three groups. f The quantification of p‐AKT (ser473) and p‐GSK3β (ser9), respectively, compared with total‐AKT and total‐GSK3β. g, h The apoptosis of HT2 cells in each group was determined by flow cytometry. i, j Representative images and quantification of western blotting showing the expression of BCL-2, Bax, and cleaved caspase 3 in different treated HT22 cells. (n = 3 per group; Values represented mean ± SD; ns indicates no significant, *P < 0.05, **P < 0.01, ***P < 0.001)
Furthermore, to determine whether hDPSCs still exert neuroprotection by activating Nrf2 via the AKT/GSK3β pathway, the expression of related gene and cell apoptosis were measured. As anticipated, compared with the GLU-treated group, hDPSCs induced the activation of p-AKT (ser473), resulting in the inhibition (increased phosphorylation) of GSK3β, accompanied by the enhanced accumulation and stabilization of Nrf2 in the nucleus (Fig. 4e, f). Then, after treating GLU-induced HT22 cells with hDPSCs, the apoptosis rate level was determined by flow cytometry, while the expression of apoptosis-related proteins was examined by western blotting. We found that the apoptosis rate of HT22 cells induced by GLU was strongly reduced after co-culture with hDPSCs (Fig. 4g, h). Not only that, but the western blot analysis also showed that hDPSCs significantly up-regulated the expression of the anti-apoptotic protein BCL2 and decreased the expression of the pro-apoptotic protein Bax and cleaved caspase 3 (Fig. 4i, j). Preincubation of LY294002 similarly confirmed that, compared to the hDPSCs-treated group, the increased protein expression of p-AKT (ser473), p-GSK3β (ser9), nuclear Nrf2, and HO-1 were reversed by LY294002 (Figure S5a–d). This inhibition of the antioxidant effect of hDPSCs ultimately led to an over-release of ROS (Figure S5e, f). In summary, the cumulative data revealed that at least to some extent, hDPSCs exert a neuroprotective effect by promoting nuclear accumulation and stabilization of Nrf2 via the p-AKT (ser473)/p-GSK3β (ser9) pathway in vitro AD cell model.
Administration of hDPSCs enhanced spatial learning and memory ability in 3xTg-AD miceIt is well known that in the onset and development of AD, whether it is oxidative stress, neuroinflammation or nerve cell dysfunction, it will eventually cause cognitive decline in AD patients. Therefore, to further explore whether hDPSCs can enhance the memory and cognitive functions of AD mice, behavioral tests including the Morris water maze (MWM) and fear conditioning test (FCT) were performed. As shown in the experimental diagram, animals were first tested in the MWM task 5 weeks after the single transplantation of hDPSCs into the bilateral hippocampus of 10-month-old 3xTg-AD mice (Fig. 5a). During the preceding 6 days of training, the AD-phosphate buffered saline (AD-PBS) mice spent more time reaching the escape platform compared with the WT and hDPSCs-treated mice (Fig. 5d). Consistent with this, the WT and hDPSCs-treated mice showed better performance in the escape latency time than that of AD-PBS mice during the test period (Fig. 5c), and the typical escape route showed a clear orientation rather than the erratic swimming path of AD-PBS mice (Fig. 5b). Moreover, after removal of the hidden platform, hDPSCs-treated mice spent longer exploration time in the target quadrant and crossed the original platform location more times within 60 s compared to the AD-PBS mice (Fig. 5e, f). In 2 days of the fear conditioning test, the freezing rate of the hDPSCs-treated mice was only slightly, albeit statistically significant, higher than that of AD-PBS mice (Fig. 5g). Astonishingly, when mouse hippocampal tissue was collected after behavioral testing, we found that the hippocampal mass of AD + PBS mice was significantly less than that of WT mice, and this difference was reversed in the hDPSCs-treated mice (Fig. 5h). These results indicated that the transplantation of hDPSCs had improved impaired cognitive function in 3xTg-AD mice.
Fig. 5
Administration of hDPSCs enhanced spatial learning and memory ability in 3xTg-AD mice. a Experimental design is illustrated schematically. b–f Results from WT-PBS, AD-PBS, and AD-DPSCs groups mice that were cognitively tested by MWM. The typical escape paths (b), the escape latency(s) (c), the time mice swam in the target quadrant (e), and the average number of crossing previous platform location in hidden‐platform test on the seventh day. d Training curves showed the average escape latency(s) to the hidden platform during the first 6 days acquisition training. g Comparison and quantification of the freezing rate (%) in the three groups of mice participating in the fear conditioning test. h Quantitative comparison of hippocampal compartments of isolated mice after behavioral testing (n = 3–10 per group; Values represented mean ± SD; ns indicates no significant, *P < 0.05, **P < 0.01)
hDPSCs reduced neuropathology in the hippocampus of 3xTg-AD miceTo determine whether improvement in cognitive function in AD mice was accompanied by changes in representative pathological features in the brain, we assessed them focusing on the mouse hippocampus. Compared to healthy individuals, patients with AD have significantly reduced brain volume, which is strongly linked to atrophy caused by synaptic degeneration and neuronal death, particularly in the vulnerable hippocampus.32,33 We systematically compared and measured the distance between the CA1 and DG regions in the hippocampus from different groups of mice at 3 different levels (S1, S2, and S3) through the NISSL-stained brain tissue slices as indicators of hippocampal atrophy.34 As shown in Fig. 6a, b, hippocampal atrophy was distinctly macroscopic in AD-PBS mice compared to WT, as confirmed by a significant reduction in S1-S3. Treatment of AD mice with hDPSCs resulted in a significant higher S2 thickness, whereas little change was detected in the other two markers. The detection of apoptosis-related genes in the hippocampus also revealed that the expression of anti-apoptotic protein (BCL2) was significantly increased, and the expression of pro-apoptotic proteins Bax and cleaved caspase 3 was decreased after hDPSCs treatment (Figure S6a, b).
Fig. 6
hDPSCs reduced neuropathology in the hippocampus of 3xTg-AD mice. a The NISSL-stained brain slice images showed arrows at S1, S2, and S3, indicating the thickness between the CA1 and DG subregions of the hippocampus. Scale = 250 μm. b Quantification of thickness for the 3 zones selected. c Aβ and AT8 immunofluorescent staining showed the extent of Aβ aggregation and Tau phosphorylation in the hippocampus between three groups mice. Scale = 20 μm. d Quantification of the Aβ- and AT8-marked areas in the hippocampus, respectively. e, f Representative images and quantification of western blotting showing the expression of APP and p-Tau (AT8) in the hippocampus between three groups mice. g, h Representative Golgi staining images and quantification of spine density (red arrows) in the hippocampus from mice. Scale = 5 μm. (n = 3 per group; Values represented mean ± SD; ns indicates no significant, *P < 0.05, **P < 0.01, ***P < 0.001)
Since the most prominent molecular pathology of AD is the neurotoxicity of aggregate Aβ and hyperphosphorylated tau protein, which jointly damage neurons and ultimately lead to cognitive decline, we performed immunofluorescence staining to assess whether hDPSCs had modified their progression.1 The results revealed that the pathological Aβ aggregation and the expression of phosphorylated Tau protein (AT8) in the hippocampus of hDPSCs-treated AD mice were much lesser than those in AD-PBS mice (Fig. 6c, d). Consistent with this, Western blotting showed that hDPSCs also reduced the protein expression of APP and p-Tau (AT8) in the hippocampus of 3xTg-AD mice (Fig. 6e, f).
We next performed Golgi staining of hippocampus to determine whether hDPSCs could affect the dendritic spines, which are the primary locus of most glutamatergic excitatory synaptic interactions within neuronal circuits.35 Consistent with the aforementioned pathologic findings, we found a significant decrease in the spine density of secondary dendritic branches of apical dendrites in CA1 regions of AD-PBS mice compared to WT mice, whereas hDPSCs treatment prevented spine loss pathology (Fig. 6g, h). Unexpectedly, we also observed that both the complexity and integrity of neurite arborization in the CA1 and DG regions of the hDPSCs-treated mice were visibly higher than those in the AD-PBS mice (Figure S7). These data together confirmed that single administrations of hDPSCs were indeed effective in improving hippocampal neuropathology in 3xTg-AD mice.
Neuroprotective efficacy of hDPSCs in 3xTg-AD mice was associated with enhancing Nrf2 nuclear accumulation via AKT/GSK3β pathwayAfter characterizing the efficacy of hDPSCs in AD models in vivo, we focused on exploring the mechanism by which hDPSCs-modulated recovery of cognitive function and neuropathological damage. Given that initial exploration of therapeutic models in vitro has shown that administration of hDPSCs improved microglial polarization and neuronal oxidative stress imbalance, which are closely linked to brain microenvironmental homeostasis such as Aβ clearance and neuronal normal communication, we expanded on these findings. By immunofluorescence, compared with AD-PBS mice, the hyperreactive microglia (IBA1+, INOS+) in the hippocampus were significantly decreased and M2 microglia marker expression (IBA1+, Arg1+) increased after 5 weeks of hDPSCs transplantation, suggesting that the administration of hDPSCs promoted the M2 polarization of microglia, which contributes to the clearance of Aβ and neuroinflammation in the AD mice brain (Figs. 7a, c, and S8a, b). DHE staining also indicated that hDPSCs remarkably diminished the ROS generation in the hippocampus of AD mice, basically consistent with normal ROS levels in WT mice (Fig. 7b, d). To determine whether engraftment of hDPSCs influences the activation of Nrf2, levels of Nrf2 and downstream targets were analyzed in the hippocampus of 3xTg-AD mice. Immunofluorescence staining of hippocampal slices revealed a significant increase in the number of Nrf2-positive cells in the hDPSCs-treated AD mice, even higher than in wild-type mice, with a marked nuclear translocation (Figure S8c, d). Western blot and quantitative analysis of hippocampal tissue also showed that the protein expression of Nrf2, HO-1, and other antioxidant-related genes in hDPSCs-treated AD mice was increased compared with that in AD-PBS mice (Fig. 7e, f).
Fig. 7
Neuroprotective efficacy of hDPSCs in 3xTg-AD mice was associated with enhancing Nrf2 nuclear accumulation via AKT/GSK3β pathway. a, c Immunofluorescence staining images and quantification of microglia (IBA1, Arg1) and IBA1-Arg1 co-localization (Merge, M2 microglia) in the hippocampus between three groups mice. Scale = 20 μm. b, d The ROS level in the hippocampus determined by DHE staining. Scale = 20 μm. e, f Representative images and quantification of western blotting showing the expression of total Nrf2, HO-1, GPX4, SOD1, and nuclear Nrf2 in different treated AD mice. g Representative western blotting results showed the expression of p-AKT (ser473) and p-GSK3β (ser9) of three groups mice in the three groups. h The quantification of p‐AKT (ser473) and p‐GSK3β (ser9), respectively, compared with total‐AKT and total‐GSK3β. (n = 3 per group; values represented mean ± SD; ns indicates no significant, *P < 0.05, **P < 0.01, ***P < 0.001)
To further determine whether the mechanism of the suppressed oxidative stress and the activation of Nrf2 in the hippocampus is consistent with in vitro models, we examined the protein expression of the AKT/GSK3β pathway. The results revealed that treatments with hDPSCs overcame these negative effects by promoting nuclear accumulation of Nrf2 via stimulating the positive effects of phosphorylation of AKT and GSK3β (Fig. 7g–h). In conclusion, we demonstrated that transplantation of hDPSCs improved the oxidative stress microenvironment in the hippocampus of 3xTg-AD mice by activating the AKT/GSK3β/Nrf2 pathway, providing excellent neuroprotective efficacy.
留言 (0)