
記住我

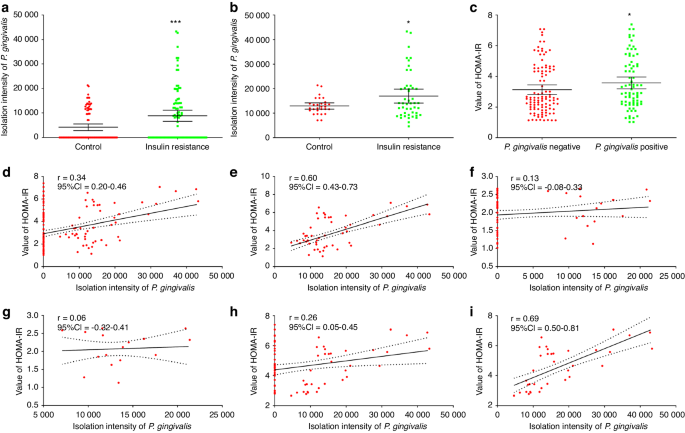


Histologically, progression from cellular atypia to varying degrees of dysplasia can ultimately lead to invasive HNSCC.2 To examine dynamic changes in the RNA modification landscape during HNSCC initiation and progression, we utilized Tgfbr1 and Pten conditional knockout (2cKO) mice, a model that develops dysplasia progressing to HNSCC upon tamoxifen administration (Figs. 1a, b and S1a).26 Liquid chromatography-tandem mass spectrometry (LC-MS/MS) screening of total RNA identified over ten RNA modifications, among which m1A levels steadily increased during cancer initiation and progression (Fig. 1c–e). We, therefore, hypothesized elevated m1A modification plays an important role in malignant transformation and immune evasion in HNSCC. To validate the LC-MS/MS screening results, we assessed m1A levels in tumors and peritumoral tissues by immunohistochemistry (IHC). Consistent with LC-MS/MS, m1A levels were significantly higher in tumor beds compared to peritumoral regions (Fig. S1b). m1A methylation is one of the most prevalent and highly conserved modifications in both eukaryotic and prokaryotic RNAs, suggesting a critical biological role. Despite unclear m1A functions in RNAs, these results suggested regulatory effects of m1A in HNSCC initiation and progression. m1A levels, modification patterns, and regulator expression might closely associate with tumorigenesis.
Fig. 1
RNA m1A modification is activated during HNSCC initiation and progression. a, b Schematic depicting LC-MS/MS assays. c The levels of various RNA modifications in total RNAs, as detected by LC-MS/MS (n = 3). d The internal m1A/A levels in total RNAs, as detected by LC-MS/MS (n = 3). e The chemical structure of m1A methylation. f Expression of genes related to m1A modification in the TCGA-HNSC dataset. g Cox regression of m1A-associated genes in the TCGA-HNSC dataset. h, i Kaplan‒Meier survival analysis of TRMT61A in the h TCGA-HNSC dataset and i GSE41613 datasets at the best cutoff values. One-way ANOVA followed by Tukey’s multiple comparisons tests (d); Mann–Whitney test (f); Cox regression (g–i); log-rank test (h, i). *P < 0.05; **P < 0.01; ***P < 0.001
TRMT61A is the key m1A regulator gene in HNSCCSimilar to dynamic m6A modification, m1A is installed by methyltransferases called “writers” (including TRMT6, TRMT61A, TRMT10C, and TRMT61B), removed by demethylases called “erasers” (including ALKBH3 and ALKBH1), and recognized by m1A-binding proteins called “readers” (including YTHDF1, YTHDF2, YTHDF3, and YTHDC1). To identify which m1A regulator contributes most to HNSCC progression, we examined the expression of m1A writers, erasers, and readers in the TCGA-HNSC cohort. mRNA levels of m1A regulators were significantly elevated in tumors compared to normal tissues, indicating activation of the m1A machinery in cancer (Fig. 1f). Among known m1A writers, TRMT61A primarily modifies cytosolic tRNAs, while TRMT61B and TRMT10C mediate m1A modification on mitochondrial RNA.27,28 TRMT61A contains a methyl donor (S-adenosyl-l-methionine, SAM) binding pocket and functions as the catalytic subunit. It forms a functional heterotetramer complex with TRMT6, which lacks the SAM binding motif but is essential for tRNA-binding.6 We found TRMT61A mRNA was elevated in both paired (Fig. S1c) and unpaired (Fig. S1d) tumor samples compared to normal samples. Further analysis revealed upregulated TRMT61A mRNA in various human cancers, including liver, breast, and colon (Fig. S2). Cox regression showed high TRMT61A expression conferred the highest hazard ratio (1.011–1.719, P = 0.0416) among these genes, while other m1A regulators were not significant (Fig. 1g). Interestingly, gene set enrichment analysis (GSEA) revealed marked activation of cell proliferation pathways and suppression of immunological pathways in TRMT61A-high tumors (Fig. S3a). Moreover, high TRMT61A predicted poor prognosis in both TCGA-HNSC and GSE41613 cohorts (Figs. 1h, i and S3b, c). Together, these findings identified TRMT61A as a key m1A regulator in cancer, warranting further study.
TRMT61A expression is associated with HNSCC progression and poor prognosisIn Tgfbr1/Pten 2cKO mice, Western blot showed spontaneous neoplastic transformation of oral mucosa was accompanied by increased TRMT61A protein levels (Fig. 2a, b). In HNSCC patients, TRMT61A expression was significantly higher in tumors versus peritumoral normal tissues (Fig. 2c, d). We investigated TRMT61A expression by IHC in HNSCC tissue microarrays comprising 42 oral mucosa, 69 dysplasia, and 210 HNSCC samples. Aligning with bioinformatic and Western blot data, IHC revealed higher TRMT61A expression in HNSCC versus dysplasia and normal mucosa (Fig. 2e, f). Further analysis demonstrated positive correlations between TRMT61A and lymph node metastasis, but not tumor size and pathologic grade (Figs. 2g and S4a, c–e). Moreover, metastatic lymph node tissue exhibited higher IHC scores than primary tumors (Fig. S4a–b). TRMT61A did not associate with other clinical parameters including age, smoking, alcohol, or radiotherapy (Table S4). Importantly, TRMT61A positively correlated with PD-L1 (Fig. 2h, i). High TRMT61A predicted poor clinical outcomes in HNSCC patients (Fig. 2j) and was an independent risk factor for overall survival (Fig. S4f, g).
Fig. 2
TRMT61A is upregulated in HNSCC tissues and predicts a poor prognosis. a, b Western blot detection (a) and normalized levels (b) of TRMT61A expression in normal oral mucosa, dysplasia, and HNSCC tissues of Tgfbr1/Pten 2cKO mice. c, d Western blot detection (c) and normalized levels (d) of TRMT61A expression in tumor and peritumoral normal tissues of HNSCC patients. e Representative IHC staining results of TRMT61A in normal mucosa, dysplasia, and HNSCC samples with different pathological grades. f Normalized TRMT61A expression in HNSCC (n = 210) compared with dysplasia (n = 69) and oral mucosa (n = 42). g Quantification of the TRMT61A histoscores among pathological grades. h Correlation of TRMT61A with other cancer-associated proteins in the HNSCC tissue microarray database. i Correlation of TRMT61A with PD-L1 in the HNSCC tissue microarray database. j Kaplan‒Meier survival analysis of TRMT61A in the HNSCC tissue microarray database at the best cutoff value. Data were mean with s.e.m. One-way ANOVA followed by Tukey’s multiple comparisons tests (b); Kruskal–Wallis test followed by Dunn’s multiple comparisons tests (f); Kruskal–Wallis test (g); two-tailed paired Student’s t-test (d); Spearman’s correlation (h, i); Cox proportional-hazards model and log-rank test (j). *P < 0.05; **P < 0.01; ***P < 0.001; ns represents no significance
To examine TRMT61A expression in the HNSCC tumor microenvironment (TME), we analyzed a single-cell sequencing dataset of 19 primary HNSCC tumors.29 Cell type annotation identified 9 populations, including cancer cells, fibroblasts, and various immune cells (Fig. S5a). TRMT61A was universally expressed across all cell types in HNSCC, with the highest levels in malignant cells and fibroblasts (Fig. S5b, c). Confocal microscopy demonstrated cytoplasmic localization of TRMT61A in CAL 27 and WSU-HN6 cells (Fig. S4h, i).
TRMT61A knockdown reduces the activity of the MYC pathwayWe screened two lentivirus-mediated TRMT61A shRNAs and selected shTRMT61A-2 (hereafter sh61A) based on superior knockdown efficiency at both mRNA and protein levels (Fig. 3a, S6a). Given previous studies suggest that TRMT61A protein promotes the dramatic transition of quiescent T cells into proliferative T cells7 and MYC is the core regulator of T cell activation,8 we hypothesize that the MYC pathway is affected upon TRMT61A knockdown. At the mRNA level, known MYC targets like CCNE1, CDK2, CD47, and CD274 were downregulated in sh61A cells (Figs. 3b, S6b). Interestingly, while MYC mRNA was unchanged, MYC protein was reduced in sh61A cells (Fig. 3c, d). Given the decrease in MYC protein, we examined MYC-regulated targets, including CDK2, cyclin E1 (encoded by CCNE1), and PD-L1 (encoded by CD274). As expected, MYC downregulation decreased cyclin E1, which regulates S phase (Fig. 3c, d and S6c). Among non-canonical targets,17 PD-L1 showed reduced mRNA and protein levels (Figs. 3b, d and S6b). Aligning with a previous report,30 MYC downregulation decreased cell size (Figs. 3e and S6f). As MYC governs stemness and epithelial-mesenchymal transition, we then examined these phenotypes in shCtrl and sh61A cells. sh61A cells exhibited over 50% lower sphere formation, indicating reduced clonogenicity (Figs. 3f and S6d). Wound healing assays revealed weaker migration and invasion in sh61A cells (Figs. 3g and S6e). Colony formation assays also showed diminished self-renewal upon TRMT61A knockdown (Fig. 3h). Consistent with the upregulated expression of TRMT61A in tumors with higher N levels and lymph metastasis (Fig. S4a–c), Western blot revealed a decrease in the expression of mesenchymal markers, N-cadherin and α-SMA, along with an increase in the expression of the epithelial markers E-cadherin and β-catenin in sh61A cells (Fig. 3i). Furthermore, the cell invasion assay demonstrated a notable reduction in the invasion capability of WSU-HN6 cells following TRMT61A knockdown (Fig. 3j). Taken together, these results provide evidence supporting the role of TRMT61A in promoting cancer stemness and epithelial-mesenchymal transition.
Fig. 3
TRMT61A promotes cancer invasion and stemness. a Knockdown of TRMT61A by shTRMT61A-2 in WSU-HN6 and CAL 27 cells were confirmed by Western blot. b mRNA levels of shCtrl/sh61A CAL 27 cells were quantified by qPCR (n = 3). c, d Protein levels of shCtrl/sh61A WSU-HN6 and CAL 27 cells were quantified by confocal microscopy (c) and Western blot (d). e Cell sizes of shCtrl/sh61A WSU-HN6 cells quantified by FSC-A (n = 3). Representative flow cytometry histograms are shown to the right. f Sphere formation ability of shCtrl/h61A CAL 27 cells (n = 3). Representative flow cytometry histograms are shown to the right. g Representative images from in vitro wound healing assays of CAL 27 cells. h Colony-forming efficacy of CAL 27 cells (left, n = 3) and WSU-HN6 cells (middle, n = 3). Representative pictures are shown to the right. i Protein levels of epithelial-mesenchymal transition indicators were quantified by Western blot. j Representative images of cell invasion assay (left) and quantitative analysis (right). k, l Analysis of high-throughput sequencing results of sh61A CAL 27 cells against shCtrl CAL 27 cells by a GSEA (k; n = 3) and a GO enrichment analysis (l; n = 3). Data were mean with s.e.m. Two-tailed unpaired Student’s t-test (b, e, f, h, j). *P < 0.05; **P < 0.01; ***P < 0.001; ns represents no significance
Cancer requires multiple genetic events and the acquisition of hallmarks of cancer.31 Importantly, MYC activation can contribute to many hallmarks, including proliferation, self-renewal, survival, genomic instability, metabolism, invasiveness, angiogenesis, and immune evasion.25 High-throughput sequencing of shCtrl and sh61A HNSCC cells revealed differentially expressed genes (Figs. S7a, S8a). Gene ontology (GO) enrichment showed involvement of differentially expressed genes in inflammatory and immune responses and cell adhesion (Fig. 3l). GSEA demonstrated marked suppression of cell metabolism and proliferation pathways, including MYC targets, cholesterol homeostasis, mTORC1 signaling, Ras signaling, reactive oxygen species, and oxidative phosphorylation in sh61A cells (Figs. 3k and S7b–d, S8b–h). Thus, TRMT6A has crucial effects in transformed cells and also enables evolving tumors to proliferate and evade the immune response.
TRMT61A promotes MYC translation but not transcriptionGiven the unchanged MYC mRNA levels and the significant decrease in MYC protein levels, we hypothesize that TRMT61A might promote MYC mRNA translation but not transcription. Translational regulation of protein synthesis occurs at initiation, elongation, and termination. tRNA decoding has a fundamental role across these steps. The human genome contains over 400 tRNA genes, with over 200 typically expressed per cell.32 tRNAs were long thought to affect translation via structures and mRNA codon interactions. However, their regulation closely relates to diverse tRNA chemical modifications like m1A.7,33 m1A deposition in tRNA is highly conserved and dependent on secondary structure, occurring across life. Cytosolic m1A sites share a GUUCNANNC sequence (A = m1A) within a hairpin, identical to tRNA T-loops.27
Considering that m1A modification is typically found at position 58 of a tRNA, we first reanalyzed the published tRNA-m1A-sequence data to demonstrate how TRMT61A knockout leads to the decrease in tRNA m1A58 modification levels on different kinds of tRNAs.7 Most tRNAs showed varying degrees of reduction in m1A58 levels upon TRMT61A knockout, and the most affected tRNAs are the ones decoding serine (TCC and AGC) and leucine (CTG and TTG) (Fig. 4a). Then we compared these tRNAs with codons of the human MYC mRNA. Interestingly, TCC, AGC, CTG, and TTG were also among the codons most frequently used by human MYC mRNA (Fig. 4b). To further examine the mechanism of MYC mRNA translation enhanced by TRMT61A, we redesigned the human MYC cDNA by replacing the codons corresponding to the tRNAs most affected by TRMT61A deletion with synonymous codons corresponding to tRNAs least affected by TRMT61A deletion (Fig. 4c). In short, TCC/AGC encoding serine and TTG/CTG encoding leucine were replaced by TCG and CTT, respectively (Fig. 4c). As expected, transfection of MYC-Mutant (MYC-Mut) plasmid in sh61A CAL 27 and WSU-HN6 cells was enough to significantly upregulate the expression of MYC in two sh61A cells in vitro (Fig. 4d). In sh61A CAL 27 cells, MYC-WT plasmid was not able to rescue the defective MYC expression. Although MYC-WT plasmids were able to slightly upregulate MYC expression in sh61A WSU-HN6 cells, MYC-Mut plasmids rescue the defective MYC expression more efficiently than MYC-WT plasmids (Fig. 4d). In shCtrl cells, both MYC-WT and MYC-Mut plasmids efficiently mediated MYC protein expression, reaching similar protein levels (Fig. 4e), indicating that the translation efficiency is similar for TCC/AGC vs. TCG and CTG/TTG vs. CTT in the presence of TRMT61A. Furthermore, both MYC-WT and MYC-Mut plasmids increase the proliferation of sh61A CAL 27 cells in vitro, demonstrating that MYC synthesized from MYC-Mut plasmids are fully functional (Fig. 4f). Together, these results of this codon-switch assay confirm that tRNA-m1A58 modification mediated by TRMT61A directly regulates translation of human MYC mRNA via codon decoding.
Fig. 4
TRMT61A promotes the translation of human MYC mRNA. a The decrease in the magnitude of the tRNA-m1A58 level in each tRNA after TRMT61A deletion. b The codon frequency of human MYC mRNA. c Schematic diagram of the human MYC codon-switch assay. d Expression of MYC-WT and MYC-Mutant (MYC-Mut) in sh61A CAL 27 and WSU-HN6 cells. Protein levels of MYC were quantified by immunoblotting (n = 4). Representative Western blot results are shown to the right. e Expression of MYC-WT and MYC-Mut in shCtrl CAL 27 and WSU-HN6 cells confirmed by Western blot. f Proliferation of sh61A CAL 27 cells transinfected with vector, MYC-Mut, and MYC-WT detected by SRB assay. Data were mean with s.e.m. Two-tailed unpaired Student’s t-test (d). One-way ANOVA followed by Tukey’s multiple comparisons tests (f). *P < 0.05; **P < 0.01; ***P < 0.001; ns represents no significance
TRMT61A promotes PD-L1 expression upon inflammationPrevious studies have shown that oncolytic virus (OV) injection induces IFNγ expression, which upregulates PD-L1 expression in cancer cells and leads to immune evasion and OV monotherapy failure.12 In Tgfbr1/Pten 2cKO mice, two OV doses significantly upregulated mRNA levels of both PD-L1 and TRMT61A (Fig. 5a, c). Interestingly, OV-treated tumors displayed elevated m1A modification levels compared to vehicle-injected tumors (Fig. 5b). Since MYC positively regulates PD-L1 expression as a transcription factor, we hypothesize TRMT61A may play a role in OV therapy cancer immune evasion by promoting reactive PD-L1 expression through enabling efficient MYC synthesis. Indeed, upon IFNγ treatment, sh61A CAL 27 and WSU-HN6 cells showed significantly reduced PD-L1 expression compared to shCtrl counterparts, while other immune checkpoint molecules like CD47 and CD155 remained unchanged (Fig. 5d, e). Given MYC also transcriptionally regulates CD47,17 we speculate negative feedback loops evolved in sh61A WSU-HN6 cells to impact CD47. Together, these data indicate PD-L1 expression increases with inflammation and TRMT61A at least partially contributes.
Fig. 5
TRMT61A promotes PD-L1 expression upon inflammation. a mRNA levels of Cd274 were quantified by qPCR (n = 6 biological replicates per group). b The internal m1A/A levels in total RNAs of tumors injected with vehicle or OV, as detected by LC-MS/MS (n = 3). c mRNA levels of Trmt61a were quantified by qPCR (n = 6 biological replicates per group). d, e Protein levels of PD-L1, CD155, and CD47 were detected by flow cytometry in shCtrl/sh61A CAL 27 (d) and WSU-HN6 (e) cells 24 h post 25 ng/mL IFNγ treatment. Representative flow cytometry results are shown at the bottom. Data were mean with s.e.m.*P < 0.05; **P < 0.01; ***P < 0.001; ns represents no significance by two-tailed unpaired Welch’s t-test
Next, we sought to explore how TRMT61A regulates the landscape of inflammation using high-throughput sequencing techniques. OV injection activates a spectrum of immune responses, among which IFNγ signaling is significantly upregulated in both injected lesions and noninjected lesions.11 At the same time, it has been reported that IFN-γ are responsible for the upregulation of PD-L1 on tumors.34 Thus, IFNγ-treated cancer cells are chosen as an in vitro model simulating an inflamed TME post-OV treatment. Upon IFNγ treatment, the MYC target genes and the IFNγ pathway genes could be classified into three subsets based on mRNA expression profiles, suggesting a regulatory program during inflammation (Fig. 6a). As observed in WSU-HN6 cells, the MYC target genes are generally downregulated in sh61A CAL 27 cells in comparison to shCtrl CAL 27 cells, with or without IFNγ treatment (Fig. 6a). Interestingly, IFNγ treatment also inhibits the transcription of most MYC target genes, indicating that proliferation is inhibited during inflammation (Fig. 6a). Reculstering of MYC target genes in IFNγ treated cells shows that genes related to cell cycle regulation, such as CDK4, CCNA2, and CKD2, are downregulated during inflammation (Fig. 6a). As excepted, the IFNγ pathway is activated upon IFN-γ treatment, leading to upregulation of immune checkpoint molecules such and CD274 and CD47 (Fig. 6a). Reculstering of IFNγ pathway genes showed a shifted expression profile in the IFNγ pathway in sh61A CAL 27 cells, with pronounced downregulation of CD274 and CD47 (Fig. 6a), reinforcing the conclusion that TRMT61A is critical to the reactive upregulation of PD-L1 on cancer cells during inflammation.
Fig. 6
TRMT61A confers resistance against oHSV. a CAL 27 expression of genes belonging to MYC target genes or the IFNγ pathway in different groups was identified according to the RNA-seq results. b, d Analysis of high-throughput sequencing results of IFNγ-treated, sh61A CAL 27 cells against IFNγ-treated, CAL 27 shCtrl cells by a GSEA (b, c; n = 3 biological replicates per group), a GO enrichment analysis (d; n = 3 biological replicates per group). e, f Results of the cytotoxicity assay for shCtrl/sh61A CAL 27 (e) or WSU-HN6 (f) cells upon oHSV infection following IFNγ pretreatment at 25 ng/mL for 24 h (n = 3 biological replicates per group). Cell viability was measured three days post infection. Representative log-transformed dose-response curves are shown to the right. Data were mean with s.e.m. Two-tailed unpaired Welch’s t-test (e, f). ***P < 0.001
GSEA of differentially expressed genes in IFNγ-treated sh61A versus shCtrl CAL 27 cells showed marked suppression of cell cycle and inflammation pathways, including G2M checkpoints, inflammation, MYC targets, E2F targets, and KRAS signaling (Figs. 6b, c and S9a-c). GO analysis revealed many were involved in immune responses like cytokine production and cell adhesion (Fig. 6d). To gain a general understanding of the impact of TRMT61A on OVs, we examined the cytotoxicity of oHSV against shCtrl and sh61A HNSCC cell lines. In both sh61A cell lines, cytotoxic assays demonstrated TRMT61A knockdown increased oHSV sensitivity, leading to a more than 90% percent decrease in 50% tissue culture infectious dose (TCID50) following oHSV treatment in comparison to their shCtrl counterparts (Fig. 6e, f).
Inhibition of TRMT61A activity elevates oncolytic viruses-induced antitumor immunityEncouraged by promising in vitro results, we speculated therapies suppressing TRMT61A activity could restore immune responses against cancers during OV therapy. Compared to OV monotherapy, the thiram and oHSV combination therapy showed the most promising antitumor effect (Fig. 7a–c). In the 4MOSC1 syngeneic oral cancer model, combination therapy eradicated tumors in two-thirds of cases by the study end, while no tumors were eradicated with oHSV alone (Fig. 7a–c). Aligning with a previous study reporting thiram as an inhibitor blocking enzymatic activity of the TRMT6/TRMT61A complex,35 thiram treatment did not significantly affect TRMT61A protein levels (Fig. S10a), whereas the levels of m1A modification were decreased in the tumors of both 4MOSC1 and Tgfbr1/Pten 2cKO HNSCC models (Fig. S10b, c).
Fig. 7
Thiram enhanced oHSV efficacy in two immunocompetent allograft mouse models. a Schematic depicting the study design for 4MOSC1 (up panel) and 4T1 (down panel) tumor inoculation and treatment with oHSV and thiram. b Macroscopic appearance of tumors from 4MOSC1 (left panel) and 4T1 (right panel) at the end of the experiment. c Tumor-growth curve of 4MOSC1 (left panel) and 4T1 tumors (right panel). d Proposed mechanism of combining oHSV and m1A inhibition. e, f Representative flow cytometric analysis images (e) and quantification (f) of CD44 and CD62L expression in the CD8+ T cells from TDLNs of 4MOSC1 tumors. g, h Representative flow cytometric analysis images (g) and quantification (h) of CD44 and CD62L expression in the CD4+ T cells from TDLNs of 4MOSC1 tumors. i, j Representative IHC images of CD3 epsilon (i) and PD-L1 (j) of 4MOSC1 tumors. Two-tailed unpaired Student’s t-test (c, f, h). Data were mean with s.e.m (n = 6 mice per group). *P < 0.05; **P < 0.01; ***P < 0.001; ns represents no significance
We speculate in immunocompetent models, TRMT61A inhibition downregulates MYC and PD-L1 simultaneously, dampening inflammation-induced immune inhibition post-oHSV (Fig. 7d). Upon MYC downregulation, loss of “don’t find me” signals enables residual tumor cell destruction and sustained regression. To test this, we examined the post-treatment immune landscape in tumor tissue, tumor-draining lymph nodes (TDLNs), and spleen by flow cytometry. T cell factor 1 (TCF-1), a Wnt pathway transcription factor, maintains CD8+ T cell stemness in the tumor microenvironment and during alloimmunity.36,37 Combination therapy significantly increased intratumoral TCF-1+ T cell fractions versus oHSV alone in a 4T1 breast cancer model (Fig. S11a–c). Beyond TCF-1, CD44 and CD62L classify T cells into central memory (Tcm), effector (Teff), and naïve subsets.38 TDLN CD8+ T cells showed increased Tcm but not Teff with combination treatment (Figs. 7e, f and S1a, b). Interestingly, in TDLN CD4+ T cells, Tcm numbers showed no significant increase, while Teff significantly increased with combination therapy (Figs. 7g, h and S12a, c). Classically activated M1 macrophages have greater antitumor effects than alternatively activated M2 types.39 Combination therapy dramatically elevated intratumoral MHCII+F4/80+ M1 macrophages versus oHSV alone (Fig. S13a–c). Mature dendritic cells (DCs) expressing CD80/86 provide secondary signals activating T cells, and combination therapy increased CD80+CD86+ DCs in 4MOSC1 TDLNs (Fig. S14a–c).
To explore the correlation between TRMT61A and PD-L1-mediated immune evasion, we evaluated the death of siTrmt61a-transfected 4MOSC1 cancer cells when co-cultured with CD8+ cells isolated from mice cured by the thiram and oHSV combination therapy (Fig. S15a–c). These CD8+ T cells exhibited substantial cytotoxicity against 4MOSC1 cells, a response that was further enhanced by the introduction of Trmt61a siRNA or PD-L1 antibodies (αPD-L1). Interestingly, the simultaneous application of both Trmt61a siRNA and αPD-L1 did not yield a significant improvement in cytotoxicity (Fig. S15c), suggesting overlapping pharmacological mechanisms. Additionally, IHC showed increased T cell infiltration with combination therapy, while oHSV-induced PD-L1 upregulation was decreased (Fig. 7i), reinforcing TRMT61A’s critical role in reactive PD-L1 upregulation during oHSV treatment. In Tgfbr1/Pten 2cKO mice, thiram treatment also dramatically reduced tumor growth rate (Fig. S16a–d). Although modest, MYC’s effects on PD-L1 expression had dramatic tumor regression consequences, consistent with minor immune regulator influences markedly impacting outcomes.17 In summary, the thiram and oHSV combination demonstrated enhanced antitumor efficacy and immune modulation versus oHSV monotherapy.
留言 (0)